
以数据为中心的方法已被用于开发用于阐明蛋白质未表征特性的预测方法;然而,研究表明,这些方法应进一步改进,以有效解决生物医学和生物技术中的关键问题,这可以通过更好地代表手头的数据来实现。新的数据表示方法主要从在自然语言处理方面取得突破性改进的语言模型中汲取灵感。最近,这些方法已应用于蛋白质科学领域,并在提取复杂的序列-结构-功能关系方面显示出非常有希望的结果。在这项研究中,土耳其中东科技大学(Middle East Technical University)的研究人员,首先对每种方法进行分类/解释,然后对它们的预测性能进行基准测试,对蛋白质表示学习进行了详细调查:(1)蛋白质之间的语义相似性,(2)基于本体的蛋白质功能,(3)药物靶蛋白家族和(4)突变后蛋白质-蛋白质结合亲和力的变化。这项研究的结论将有助于研究人员将基于机器/深度学习的表示技术应用于蛋白质数据以进行各种预测任务,并激发新方法的发展。该研究以「Learning functional properties of proteins with language models」为题,于 2022 年 3 月 21 日发布在《Nature Machine Intelligence》。

蛋白质科学是一门广泛的学科,它通过实验室实验(即蛋白质组学)和计算方法(例如分子建模、机器学习、数据科学)分析单个蛋白质以及生物体的整个蛋白质组,最终创建准确且可重复使用的方法用于生物医学和生物技术。蛋白质信息学可以定义为蛋白质科学的计算和以数据为中心的分支,通过它对蛋白质的定量方面进行建模。蛋白质的功能表征对于开发新的有效的生物医学策略和生物技术产品至关重要。截至 2021 年 5 月,UniProt 蛋白质序列和注释知识库中约有 2.15 亿条蛋白质条目;然而,其中只有 56 万份(约 0.26%)由专家手动审查和注释,这表明当前的排序(数据生产)和注释(标签)能力之间存在很大差距。这种差距主要是由于从湿实验室实验及其手动管理中获得结果的成本较高,同时具有时间密集性。为了补充基于实验和管理的注释,使用计算机方法势在必行。在这种情况下,许多研究小组一直致力于开发新的计算方法来预测蛋白质的酶活性、生物物理特性、蛋白质和配体相互作用、三维结构以及最终的功能。蛋白质功能预测(PFP)可以定义为自动或半自动地将功能定义分配给蛋白质。生物分子功能的主要术语被编入基因本体论(GO)系统;这是一个概念的分层网络,用于注释基因和蛋白质的分子功能,以及它们的亚细胞定位和它们所涉及的生物过程。PFP 最全面的基准项目是功能注释的关键评估(CAFA)挑战;在该项目中,参与者预测一组目标蛋白的基于 GO 的功能关联,这些目标蛋白的功能后来通过手动调节确定,用于评估参与预测因子的性能;迄今为止的 CAFA 挑战表明,PFP 仍然是一个开放的问题。以前的研究已经表明,复杂的计算问题,其中特征是高维的并且具有复杂/非线性关系,适合基于深度学习的技术。这些技术可以有效地从嘈杂的高维输入数据中学习与任务相关的表示。因此,深度学习已成功应用于计算机视觉、自然语言处理和生命科学等各个领域。生物分子的特征(例如,基因、蛋白质、RNA 等)应被提取并编码为定量/数值向量(即表示),以用于基于机器/深度学习的预测建模。给定生物分子的原始和高维输入特征,表示模型将该特征向量计算为该生物分子的简洁和正交表示。经过优化训练的监督预测系统可以有效地学习数据集中样本的特征,并使用这些表示作为输入来执行预测任务(例如,序列上的 DNA 结合区域、生化特性、亚细胞定位等)。蛋白质表示方法可以分为两大类;(1)经典表示(即模型驱动的方法),使用预定义的属性规则生成,例如基因/蛋白质之间的进化关系或氨基酸的物理化学性质,以及(2)数据驱动的表示,使用统计和机器学习算法(例如人工神经网络)构建,这些算法针对预定义任务进行训练,例如预测序列上的下一个氨基酸。之后,训练模型的输出——即表示特征向量——可以用于其他与蛋白质信息学相关的任务,例如功能预测。从这个意义上说,表示学习模型利用了知识从一个任务到另一个任务的转移。这个过程的广义形式被称为迁移学习,据报道它在时间和成本方面是一种高效的数据分析方法。因此,蛋白质表示学习模型最大限度地减少了对数据标记的需求。蛋白质表示学习是一个年轻但高度活跃的研究领域,主要受到自然语言处理 (NLP) 方法的启发。因此,蛋白质表示学习方法在文献中经常被称为蛋白质语言模型。之前的研究表明,各种蛋白质表示学习方法,尤其是那些结合了深度学习的方法,已经成功地提取了蛋白质的相关固有特征。参见:https://www.nature.com/articles/s42256-022-00457-9/tables/1尽管有研究评估学习的蛋白质表示模型,但需要进行全面的调查和基准测试,以便在学习蛋白质的多个方面(包括基于本体的功能定义、语义关系、家族和相互作用)的背景下系统地评估这些方法。在新的研究中,中东科技大学的研究人员对自 2015 年以来提出的可用蛋白质表示学习方法进行了全面调查,并通过详细的基准分析测量了这些方法捕获蛋白质功能特性的潜力。涵盖了经典和基于人工学习的方法,并深入了解了它们各自代表蛋白质的方法。研究人员根据它们的技术特征和应用对这些方法进行分类。为了评估每个表示模型在多大程度上捕获了功能信息的不同方面,该团队构建并应用了基于以下的基准:(1) 蛋白质之间的语义相似性推断,(2) 基于本体的 PFP,(3) 药物靶蛋白家族分类,(4) 蛋白质-蛋白质结合亲和力估计。
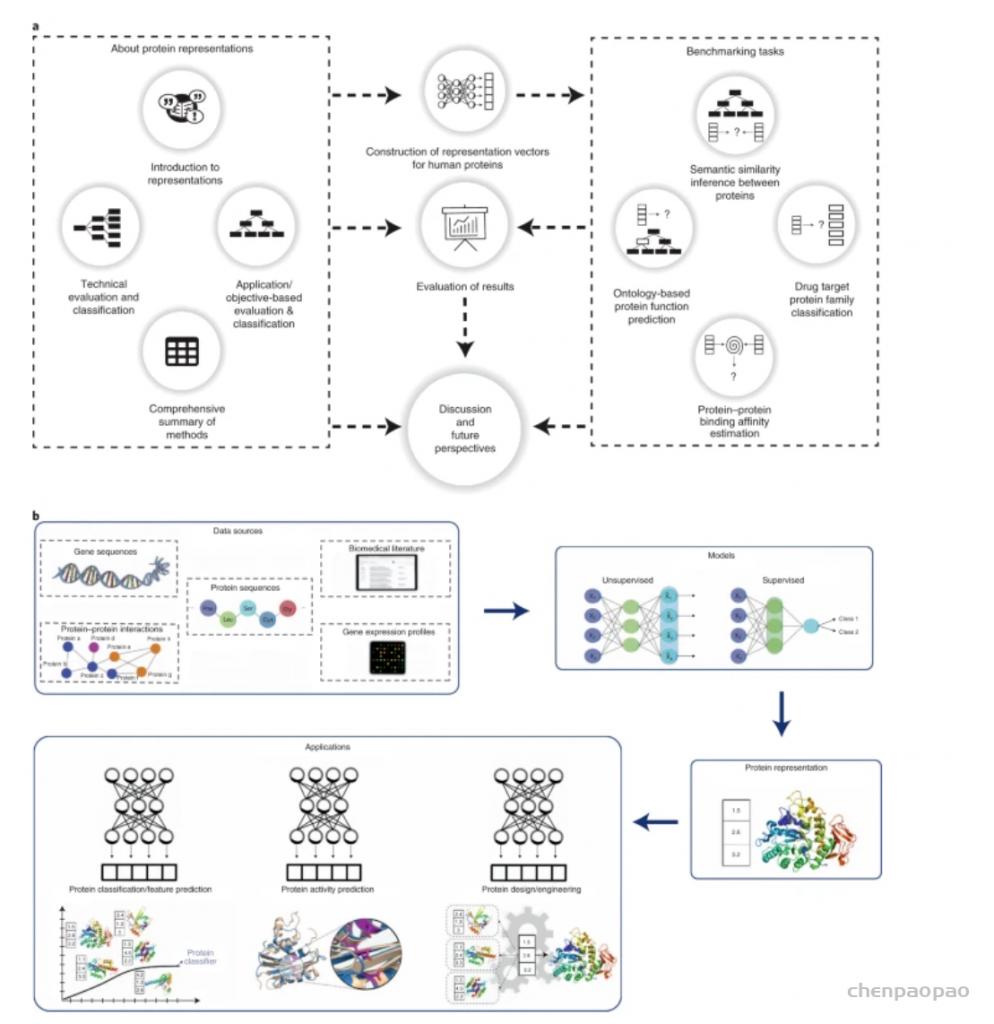
图示:研究的示意图。(来源:论文)此外,该团队还提供了相关的基准测试软件(Protein Representation Benchmark, PROBE),它允许人们轻松评估任何表示方法在该团队定义的四个基准测试任务中的性能。研究人员希望该工作能够,为希望将基于机器/深度学习的表示技术,应用于生物分子数据进行预测建模的研究人员提供信息。也希望这项研究能够激发新的想法,以开发新颖、复杂和强大的以数据为中心的方法来解决蛋白质科学中的开放问题。基于表示学习的方法在蛋白质功能分析中的表现通常优于经典方法在该团队所有的基准测试中,观察到学习表示(尤其是大型模型)在预测性能方面优于经典模型,证实了基于人工学习的数据驱动方法在表示生物分子的功能特性方面的优势。另一方面,在 PFP 预测基准的分子功能类别中,HMMER 是一种基于隐马尔可夫模型(HMM)的生物分子相似性检测和功能注释的经典方法,可以与基于深度学习的蛋白质表示方法竞争。该结果与先前的研究一致,即序列相似性与蛋白质的生化特性高度相关,以至于使用此特征的简单矢量表示几乎可以执行复杂的序列建模方法。鉴于这些结果,研究人员表示,将同源信息明确纳入表征学习模型的训练可能会导致考虑到预测性能的改进。这从基于深度学习的高性能蛋白质结构预测器(例如 RoseTTAFold 和 AlphaFold2)中也很明显,它们使用多个序列比对来显着丰富基于序列的输入。他们认为,在当前状态下,学习到的蛋白质表示对于其他原因也是必不可少的。模型设计和训练数据类型/来源是表征学习的关键因素蛋白质表征学习中最关键的因素之一是表征模型的设计。例如,在这里的基准测试中,包含了两种类型的 BERT 模型。TAPE-BERT-PFAM 接受了 3200 万个蛋白质结构域序列的训练。ProtBERT-BFD 训练有 21 亿个宏基因组序列片段;然而,这两者之间的性能差异是微不足道的。另一方面,使用相同 2.1B 数据集(例如 ProtT5-XL)训练的更复杂的模型在大多数基准测试中表现出更好的性能。因此,研究人员认为模型设计/架构是最重要的(与这些方法的设计/架构相关的信息在方法中给出,并在结果部分就预测性能进行了讨论)。关于训练数据源的另一个发现是,合并多种数据类型可能会在与功能相关的预测任务中带来更好的性能。例如,AAC 和 APAAC 都使用氨基酸组成;然而,APAAC 还在其表示模型中添加了物理化学特性,并且在语义相似性推断和 PFP 基准测试中表现得更好。同样,Mut2Vec 结合了突变配置文件、PPI 和文本数据,并取得了最佳性能;尤其是在语义相似性推理基准测试中。在蛋白质表征学习方法的构建和评估过程中应考虑潜在的数据泄漏数据泄漏可以定义为机器学习方法的训练和验证阶段之间的知识意外泄漏,导致性能测量过于乐观,是性能测试期间应考虑的关键问题。研究人员分析发现,某些表示模型在与这些模型预训练的任务生物学相关的任务中表现良好;尽管数据和实际任务彼此不同。蛋白质表征学习的现状和挑战蛋白质表示学习领域存在一些挑战。尽管大多数蛋白质表示学习模型(迄今为止提出的)都是源自 NLP 模型(基于 LSTM/transformer 的深度学习模型),但建模语言和蛋白质的问题之间存在结构差异。据估计,一个以美国为母语的成年英语使用者,平均使用 46,200 个词条和多词表达;然而,蛋白质中只有 20 种不同的氨基酸,它们被表示模型以类似于语言的引理的方式处理。这些 NLP 模型为每个单词计算一个表示向量。类似地,当这种方法应用于蛋白质序列数据时,会为每个氨基酸计算一个表示向量。这些向量被汇集起来,为每个句子/文档和蛋白质创建固定大小的向量,分别用于 NLP 和蛋白质信息学任务。因此,与 NLP 相比,蛋白质表示中的少量构建块(即 20 个氨基酸)可能为较小的模型在与蛋白质表示学习领域中的较大模型竞争时带来优势。因此,鼓励对蛋白质序列特异性学习模型进行更多研究。
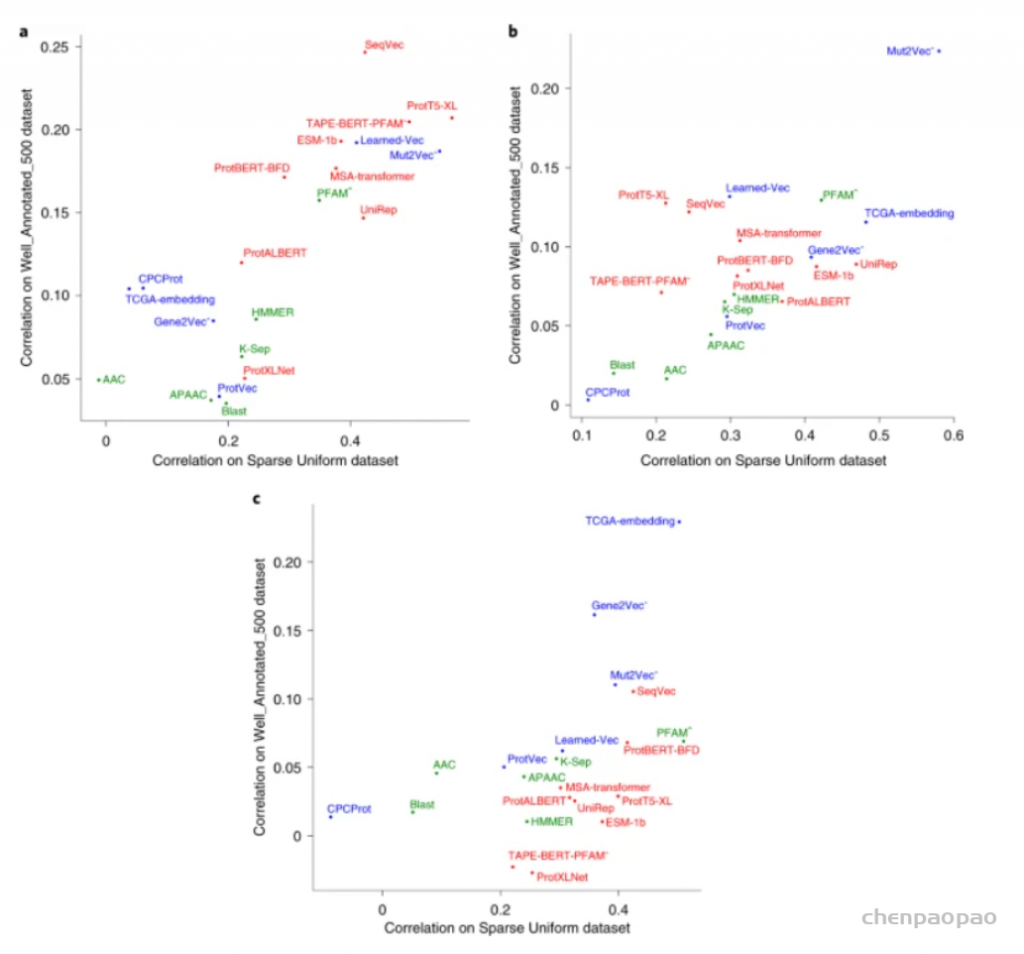
图示:蛋白质语义相似性推理基准结果。(来源:论文)模型的可解释性对于理解模型为何如此行事至关重要。在可解释表示中,所有特征都以隔离形式编码,这意味着向量上每个位置对应的特征是已知的;然而,该研究中研究的大多数学习蛋白质表示是不可解释/可解释的。例如,蛋白质中 TIM 桶结构的存在可能在其表示向量的第五位编码,而分子量信息可能在第三和第四位之间共享。一般来说,在数据科学领域,解缠结研究试图将样本的真实属性与输出向量的各个位置联系起来。蛋白质表征的解开是一个新课题,迄今为止只有少数表征模型开发人员探讨了这个问题。因此,尚不存在系统方法,并且需要新的框架来标准化评估蛋白质表示模型的可解释性。迄今为止提出的大多数蛋白质表示模型仅使用一种类型的数据(例如蛋白质序列)进行训练。然而,蛋白质知识与多种类型的生物信息相关,例如 PPI、翻译后修饰、基因/蛋白质(共)表达等;只有少数可用的蛋白质表示模型使用了多种类型的数据。在该团队的基准研究时所涉及的方法中,Mut2Vec 就是一个这样的例子,它结合了 PPI、突变和生物医学文本,并且比 GO BP 和基于 CC 的 PFP 中的许多仅基于序列的表示产生了更准确的结果。研究人员建议整合其他类型的蛋白质相关数据,尤其是进化关系,可能会进一步提高预测任务的准确性。MSA-Transformer 和无向图模型(例如 DeepSequence)通过深度学习利用同源信息。DeepSequence 使用 MSA 的后验分布计算潜在因子,而 MSA-Transformer 使用基于行和列的注意力来结合 MSA 和蛋白质语言模型。尽管 MSA-Transformer 在基准测试中表现出平均性能,但在之前的文献中,发现它在二级结构和接触预测任务上是成功的,这表明 MSA-Transformer 具有捕捉进化关系的能力。与此相关的是,文献中明确要求能够从广义的角度有效地表示蛋白质的整体蛋白质载体,用于各种不同的蛋白质信息学相关目的。研究人员认为,可以通过连接多个先前使用不同类型的生物数据独立构建的表示向量来创建这些整体表示,并使用这些向量的集成版本训练新模型以用于高级监督任务,例如预测生物过程和/或复杂的结构特征。构建这些整体表示的另一种方法是通过图表示学习直接学习整合多种蛋白质关系(例如,其他蛋白质、配体、疾病、表型、功能、途径等)的异构图。
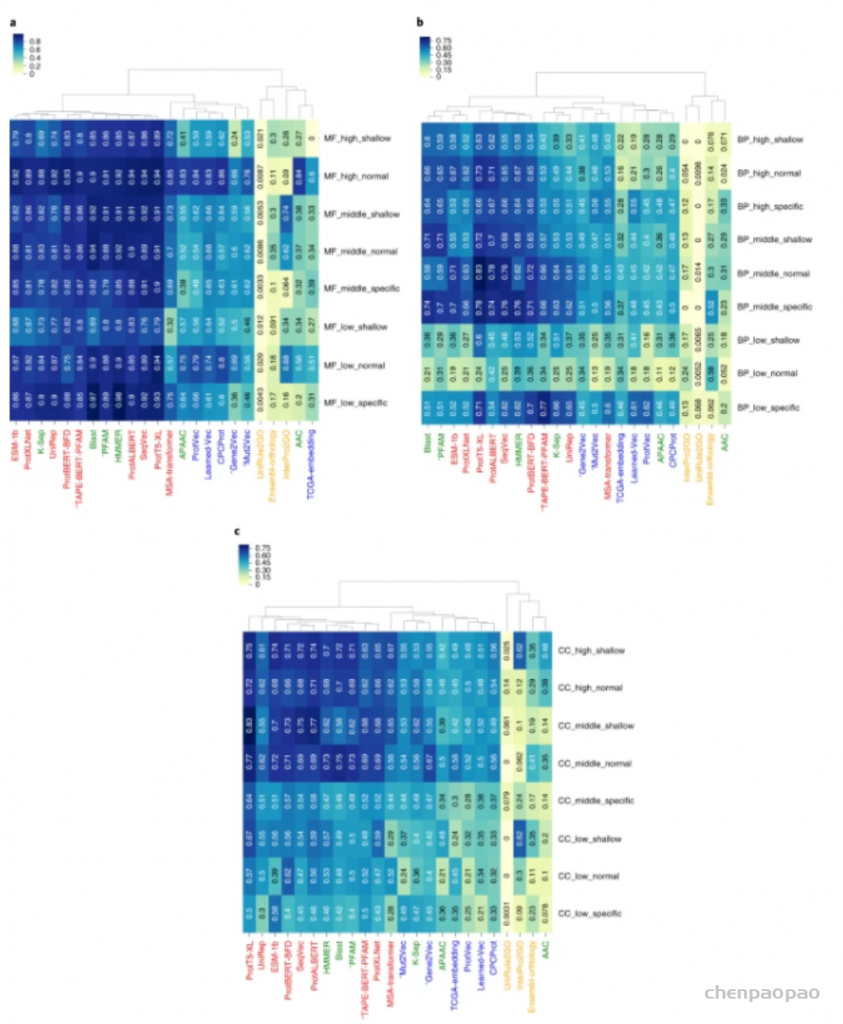
图示:基于本体的蛋白质功能预测基准结果。(来源:论文)蛋白质表示学习方法可用于设计新蛋白质蛋白质设计是生物技术的主要挑战之一。合理的蛋白质设计涉及评估许多不同替代序列/结构的活性和功能,以为实验验证提供最有希望的候选者,这可以看作是一个优化问题。为此目的要探索的序列空间是巨大的。例如,人类蛋白质的平均长度约为 350 个氨基酸,其中存在 20^350 种不同的组合,尽管其中大多数是非功能性序列。在过去的几十年中,计算方法已被用于蛋白质设计,并且这些方法已经产生了有希望的结果,特别是在酶设计、蛋白质折叠和组装以及蛋白质表面设计方面。因此已经开发出高效的抗体和生物传感器。其中一些方法使用量子力学计算、分子动力学和统计力学,每种方法的计算成本都非常高,并且需要专业知识。类似的缺点也表现在主流的蛋白质设计软件,如 Rosetta。最近的研究表明,基于人工学习的生成建模可用于从头蛋白质设计。在机器学习领域,生成建模与判别建模相反,是一种生成合成样本的方法,这些样本服从从真实样本中学习到的概率分布。这是通过有效地学习训练数据集中样本的表示来实现的。
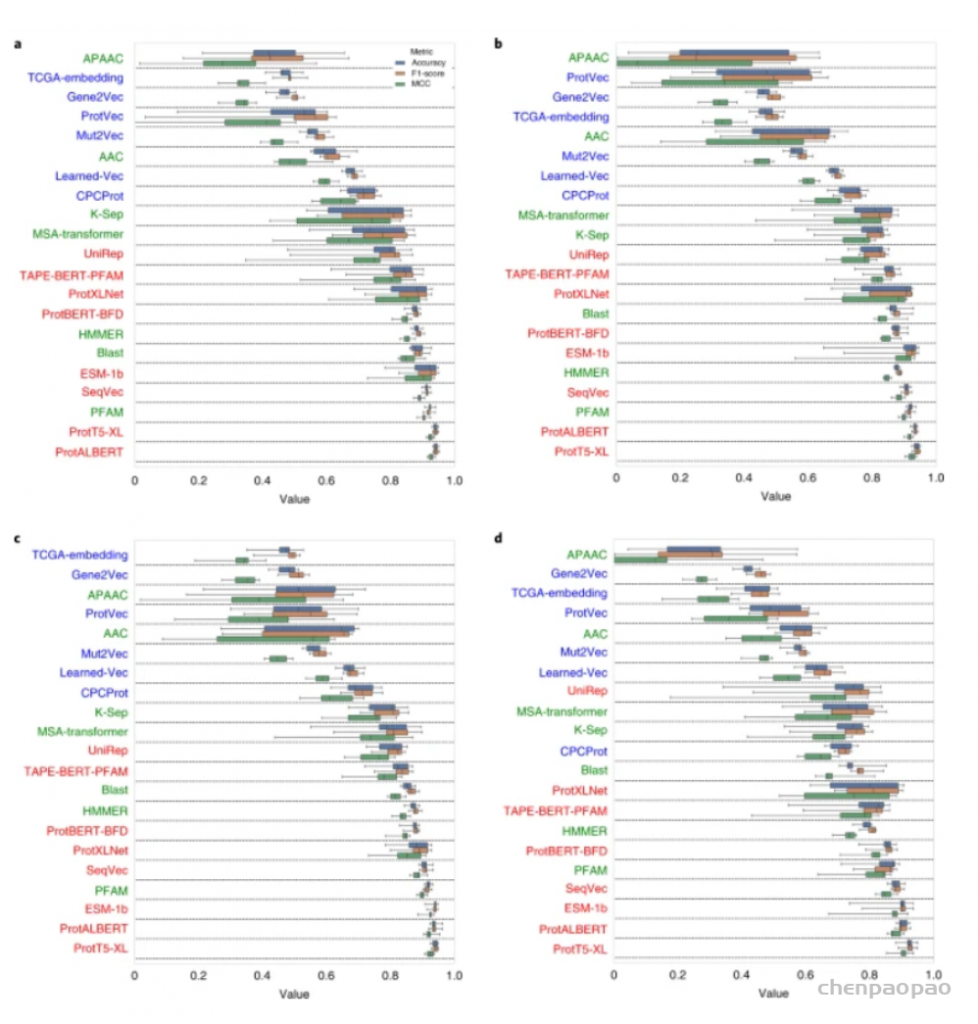
图示:药物靶蛋白家族分类基准结果。(来源:论文)深度学习最近已成为生成模型架构的关键方法,并已应用于包括蛋白质/肽设计在内的各个领域。例如,Madani 团队使用蛋白质语言模型从头开始设计属于不同蛋白质家族的新功能蛋白质,并通过湿实验室实验验证了他们的设计。这些研究表明,表示学习对于蛋白质和配体(药物)设计中的新应用至关重要。
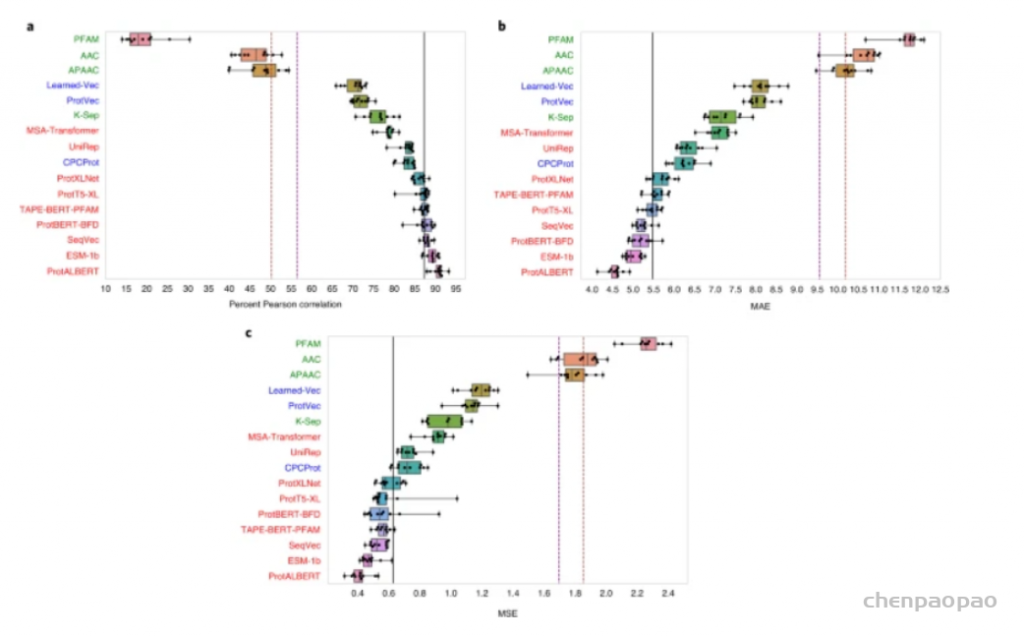
图示:蛋白质-蛋白质结合亲和力估计基准结果。(来源:论文)研究人员相信蛋白质表示学习方法将在不久的将来对蛋白质科学的各个领域产生影响,并在现实世界中应用,这要归功于它们在输入级别集成异构蛋白质数据(即理化性质/属性、功能注释等)的灵活性,以及它们有效提取复杂潜在特征的能力。论文链接:https://www.nature.com/articles/s42256-022-00457-9