

https://www.nature.com/articles/s41586-021-03819-2
https://github.com/deepmind/alphafold
论文补充材料:https://www.biorxiv.org/content/10.1101/2021.10.04.463034v1
摘要
蛋白质对生命至关重要,了解它们的结构可以促进对其功能的系统理解。通过大量的实验,已经确定了大约 100,000 种独特的蛋白质结构,但这仅代表了数十亿已知蛋白质序列中的一小部分。
蛋白质结构是指蛋白质分子的空间结构。作为一类重要的生物大分子,蛋白质主要由碳、氢、氧、氮、硫等化学元素组成。所有蛋白质都是由20种不同的L型α氨基酸连接形成的多聚体,在形成蛋白质后,这些氨基酸又被称为残基。
蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。
蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。
蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。
蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
仅根据其氨基酸序列预测蛋白质的三级结构,这是“蛋白质折叠问题”, 50 多年来一直是一个重要的开放性研究问题。尽管最近取得了进展,但现有方法仍远未达到原子级准确度,尤其是当没有可用的同源结构时。
同源结构,那些不同物种因来自共同祖先而具有的相似性结构。 例如现代马经较长时间的修饰成为具有一个趾,鼹鼠及其他洞穴动物成为瘤状肢体,大象的肢体成为柱状,这样功能各异的前肢有一个共同来源,他们都来自原始陆生脊椎动物五趾型的肢体。
在这里,我们提供了第一种计算方法,即使在不知道相似结构的情况下,它也可以以原子精度定期预测蛋白质结构。我们在具有挑战性的第 14 次蛋白质结构预测关键评估 (CASP14) 中验证了我们基于神经网络模型的完全重新设计的AlphaFold,在大多数情况下表现出与实验相媲美的准确性,并且大大优于其他方法。支持最新版本的 AlphaFold 是一种新颖的机器学习方法,它将关于蛋白质结构的物理和生物学知识,利用多序列比对,融入深度学习算法的设计中。
序列比对指将两个或多个序列排列在一起,标明其相似之处。序列中可以插入间隔(通常用短横线“-”表示)。对应的相同或相似的符号(在核酸中是A, T(或U), C, G,在蛋白质中是氨基酸残基的单字母表示)排列在同一列上。
tcctctgcctctgccatcat—caaccccaaagt
|||| ||| ||||| ||||| ||||||||||||
tcctgtgcatctgcaatcatgggcaaccccaaagt
多序列比对是成对比对的延伸,是为了在一次比对里面处理多于两条的的序列。多序列比对方法试图比对一个指定序列集合里面的所有序列,这可以帮助确定这些序列的共同区段。进行多序列比对有几种方法,最常用的一种是Clustal程序集,它使用渐进多序列比对算法。Clustal在cladistics中被用来建立进化树,在PSI-BLAST和Hidden Markov model (HMM)中用来建立序列档案以在序列数据库中搜索更远的同源序列。
从蛋白质序列预测蛋白质3D结构的计算方法的发展沿着两条互补的路径前进,分别关注物理相互作用或进化历史。 物理相互作用方案将我们认知的分子驱动力(molecular driving forces)整合到物理热力学或动力学模拟或统计模型逼近中。 虽然理论上非常吸引人,但由于分子模拟的计算难度、蛋白质稳定性的上下文依赖性以及难以产生足够准确的蛋白质物理学模型,这种方法已被证明对即使是中等大小的蛋白质也极具挑战性。 近年来,进化方案提供了一种替代方案,其中蛋白质结构约束来自蛋白质进化历史的生物信息学分析、与已解决结构的同源性和成对进化相关性。
这种生物信息学方法极大地受益于蛋白质数据库 (PDB) 中存储的实验蛋白质结构的稳定增长、基因组测序的爆炸式增长以及用于解释这些相关性的深度学习技术的快速发展。 尽管取得了这些进展,基于物理和进化历史的方法产生的预测远低于实验准确性。
我们(DeepMind团队)开发了第一个能够在大多数情况下预测蛋白质结构接近实验准确性的计算方法。 我们开发的神经网络 AlphaFold 已进入 CASP14 评估(2020 年 5 月至 7 月)。 CASP 评估每两年进行一次,使用未在 PDB 中存放或公开披露的最近解决的结构,因此它是对参与方法的盲测,长期以来一直作为结构预测准确性的金标准评估。
蛋白质结构预测 (CASP) 实验的批判性评估旨在建立蛋白质结构预测的当前技术水平,确定已取得的进展,并突出未来可能最有成效的工作重点。
… 省略了部分内容
在图 2a 中证明,CASP14 中展示AlphaFold的高准确率扩展到最近大量PDB结构样本,其中所有结构在我们的训练数据截止后都存储在PDB中,并作为完整链进行分析。此外,当主链预测准确时,观察到高侧链准确性(图 2b),并且表明我们预测的局部距离差异测试(pLDDT)置信度可靠地预测了 Ca 局部距离差异测试(lDDT-Cα)准确度相应的预测(图 2c)。我们还发现可以准确估计全局叠加度量模板建模分数 (TM-score)(图 2d)。总体而言,这些验证了 AlphaFold 在 CASP14 蛋白上的高精度和可靠性也可以迁移到最近 PDB 提交的未经整理的数据集中。
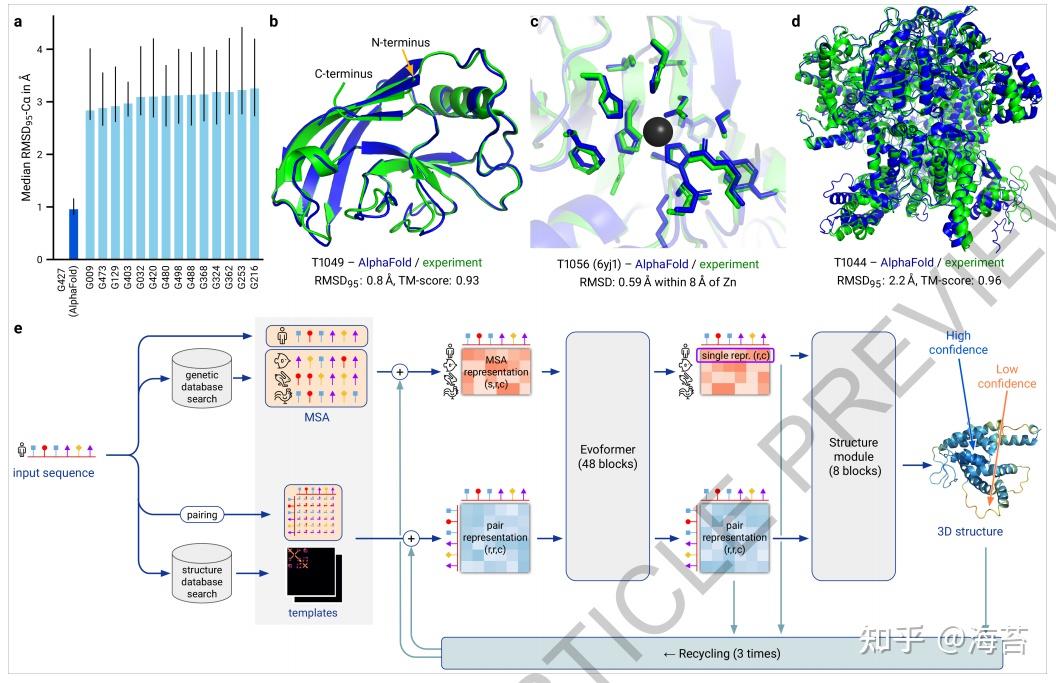
Fig. 1 | AlphaFold produces highly accurate structures.
PDB蛋白质结构数据库(Protein Data Bank,简称PDB)是美国Brookhaven国家实验室于1971年创建的,由结构生物信息学研究合作组织(Research Collaboratory for Structural Bioinformatics,简称RCSB)维护。和核酸序列数据库一样,可以通过网络直接向PDB数据库提交数据。
AlphaFold网络
AlphaFold 通过结合基于蛋白质结构的进化、物理和几何约束的新型神经网络架构和训练程序,大大提高了结构预测的准确性。特别是,我们展示了一种联合嵌入多序列比对 (MSA) 和成对特征的新架构、一种新的输出表示和相关损失函数,可实现准确的端到端结构预测、新的等变注意架构、使用中间损失函数来实现预测的迭代改进,屏蔽 MSA 损失与结构联合训练,使用自蒸馏从未标记的蛋白质序列中学习,以及自我估计准确率。
蒸馏,就是知识蒸馏,将教师网络(teacher network)的知识迁移到学生网络(student network)上,使得学生网络的性能表现如教师网络一般;或者大型模型迁移到小型模型中,小型模型的参数规模小,运行速度快,但性能与大型模型参不多。
AlphaFold 网络使用一级氨基酸序列和同源物的比对序列作为输入,直接预测给定蛋白质的所有重原子的 3-D 坐标(图 1e,请参阅方法了解输入的详细信息,包括数据库、MSA 构建和使用的模板)。 最重要的方法和组件的描述如下在附件中提供了完整的网络架构和训练过程在附件的方法章节中。
该网络包括两个主要阶段。 首先,网络的主干通过我们称为 Evoformer 的新型神经网络块的重复层处理输入,以生成 Nseq × Nres 数组(Nseq:序列数,Nres:残基数),表示已处理的 MSA 和 Nres × Nres 数组,表示残基对。 MSA 表示是用原始 MSA 初始化的,但请参阅 附件-方法 1.2.7 了解处理非常深的 MSA 的详细信息。 Evoformer 块包含许多新颖的基于注意力和非基于注意力的组件。 我们在“可解释性”部分展示了证据,表明在 Evoformer 块中早期出现了具体的结构假设并不断完善。 Evoformer 模块的关键创新是在 MSA 内交换信息的新机制和允许直接推理空间和进化关系的配对表示。
网络的主干之后是结构模块,该模块以蛋白质的每个残基(全局刚体框架)的旋转和平移的形式引入了明确的 3-D 结构。这些表示在简单的状态下初始化,所有旋转设置为一致,所有位置设置为原点,但快速发展和完善,具有精确原子细节的高度准确的蛋白质结构。网络这一部分的关键创新包括打破链原子结构以允许同时对结构的所有部分进行局部细化,一种新颖的等变变换器允许网络隐式推理未表示的侧链原子,以及一个损失项代替残基的方向正确性的重要权重。在结构模块和整个网络中,我们通过反复将最终损失函数应用于输出,然后将输出递归地提供给相同的模块来强化迭代细化的概念。使用整个网络的迭代细化(我们称之为“循环”) 对准确性有显着贡献,而额外的训练时间很少(有关详细信息,请参阅附件-方法-1.8)。
Evoformer模块
名为 Evoformer(图 1e 和 3a)的网络构建块的关键原理是将蛋白质结构预测视为 3-D 空间中的图推理问题,其中图的边缘由邻近的残基定义。 配对表示的元素编码有关残基之间关系的信息(图 3b)。 MSA 表示的列编码输入序列的各个残基,而行表示这些残基出现的序列。 在这个框架内,我们定义了许多更新操作,这些更新操作应用于每个块中,其中不同的更新操作被串联应用。
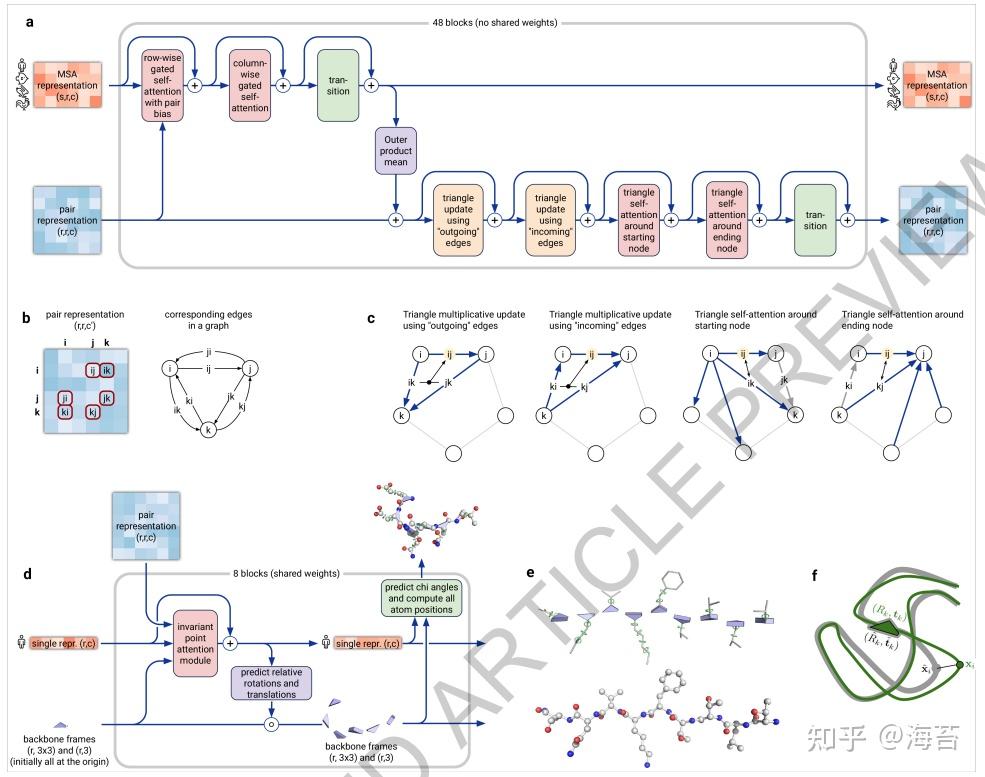
Fig. 3 | Architectural details.
MSA 表示通过在 MSA 序列维度上求和的逐元素外积更新配对表示。 与之前的工作不同,此操作应用于每个块中,而不是在网络中应用一次,这使得从不断发展的 MSA 表示到配对表示的连续通信成为可能。
在配对表示中,有两种不同的更新模式。两者都受到配对表示一致性必要性的启发——为了将氨基酸的配对描述表示为单个 3-D 结构,必须满足许多约束,包括距离上的三角不等式。基于这种直觉,我们根据涉及三个不同节点的边三角形来安排对表示的更新操作(图 3c)。特别是,我们向轴向注意力添加了一个额外的 logit 偏差,以包括三角形的“缺失边”,并且我们定义了一个非注意力更新操作“三角形乘法更新”,它使用两条边来更新缺失的第三条边(参见 附件-方法-1.6.5 了解详情)。三角形乘法更新最初是作为注意力更对称且更便宜的替代品而开发的,仅使用注意力或乘法更新的网络都能够产生高精度结构。然而,两个更新的组合更准确。
我们还在 MSA 表示中使用了一种轴向注意力的变体。 在 MSA 中的 per-sequence attention 期间,我们从 pair stack 中投射额外的 logits 以偏置 MSA attention。 这通过提供从配对表示返回到 MSA 表示的信息流来关闭循环,确保整个 Evoformer 模块能够完全混合对和 MSA 表示之间的信息,并为结构模块中的结构生成做好准备。
端到端结构预测
结构模块(图 3d)使用对表示和来自主干的 MSA 表示的原始序列行(“single representation”,“单一表示”)在具体的 3-D 主干结构上运行。 3-D 主干结构表示为 Nres 独立的旋转和平移,每个旋转和平移相对于全局框架(residue gas,“残余气”?,图 3e)。这些旋转和平移,代表 N-Cα-C 原子的几何形状,优先考虑蛋白质骨架的方向,以便每个残基的侧链位置在该框架内受到高度限制。相反,肽键几何形状完全不受约束,并且在应用结构模块期间观察到网络经常违反链约束,因为打破此约束允许对链的所有部分进行局部细化,而无需解决复杂的闭环问题。在微调期间通过违规损失项鼓励满足肽键几何形状。只有在 Amber力场中的梯度下降结构的预测后松弛中才能实现肽键几何形状的精确执行。根据经验,这种最终松弛不会提高模型的准确性,如通过全局距离测试 (GDT) 或 IDDT-Cα34 测量的,但确实消除了分散注意力的立体化学违规而不会损失准确性。
AMBER力场是在生物大分子的模拟计算领域有着广泛应用的一个分子力场。AMBER力场的优势在于对生物大分子的计算,其对小分子体系的计算结果常常不能令人满意。
residue gas表示分两个阶段迭代更新(图 3d)。首先,我们称为不变点注意力 ( Point Attention) 的新型几何感知注意操作用于更新 Nres 神经激活集(single representation,“单一表示”)而不改变 3-D 位置,然后对residue gas使用更新的激活。不变点注意力通过在每个残基的局部框架中产生的 3-D 点来增强每个通常的注意力查询、键和值,这样最终值对全局旋转和平移是不变的(参见方法“不变点注意力(IPA)”了解详情)。 3-D 查询和键也对注意力施加了强烈的空间/局部性偏差,这非常适合蛋白质结构的迭代细化。在每个注意力操作和逐元素转换块之后,该模块计算每个主干帧的旋转和平移的更新。这些更新在每个残差的局部框架内的应用使得整体注意力和更新块成为对residue gas的等变操作。
侧链 chi 角的预测以及结构的最终每个残基精度 (pLDDT) 是在网络末端的最终激活上使用小的每个残基网络计算的。 TM 分数 (pTM) 的估计值是从成对错误预测中获得的,该预测被计算为最终对表示的线性投影。最后的损失(我们称之为帧对齐点误差(FAPE)(图 3f))将预测的原子位置与许多不同对齐下的真实位置进行比较。对于每个对齐,通过将预测帧 (Rk,tk) 对齐到相应的真实帧来定义,我们计算所有预测原子位置 xi 与真实原子位置的距离。由此产生的 Nframes × Natoms 距离受到限制的 L1 损失的惩罚。这对原子相对于每个残基的局部框架是正确的产生了强烈的偏见,因此在其侧链相互作用方面是正确的,并为 AlphaFold 提供了手性的主要来源(增刊-方法 1.9.3 和增刊-图 9)
使用标记和未标记数据进行训练
AlphaFold 架构能够仅使用对 PDB 数据的监督学习来训练到高精度,但我们能够使用类似于noisy-student自我蒸馏的方法来提高准确性(见图 4a)。 在这个过程中,我们使用一个训练有素的网络来预测来自 Uniclust30的约 350,000 个不同序列的结构,并将预测结构的新数据集过滤为高置信度子集。 然后,我们使用 PDB 和这个新的预测结构数据集的混合作为训练数据从头开始训练相同的架构,其中各种训练数据增强(例如裁剪和 MSA 子采样)使网络难以重现先前预测的结构。 这种自蒸馏过程有效地利用了未标记的序列数据,并显着提高了所得网络的准确性。
Self-training是最简单的半监督方法之一,其主要思想是找到一种方法,用未标记的数据集来扩充已标记的数据集。算法流程如下:
(1)首先,利用已标记的数据来训练一个好的模型,然后使用这个模型对未标记的数据进行标记。
(2)然后,进行伪标签的生成,因为我们知道,已训练好的模型对未标记数据的所有预测都不可能都是好的,因此对于经典的Self-training,通常是使用分数阈值过滤部分预测,以选择出未标记数据的预测标签的一个子集。
(3)其次,将生成的伪标签与原始的标记数据相结合,并在合并后数据上进行联合训练。
(4)整个过程可以重复n次,直到达到收敛。
此外,我们随机屏蔽或突变 MSA 中的单个残基,并具有来自 Transformers (BERT) 式目标的双向编码器表示来预测 MSA 序列的屏蔽元素。 这个目标鼓励网络学习解释系统发育和协变关系,而无需将特定的相关统计量硬编码到特征中。 与最近的独立工作相比,BERT 目标是在相同的训练示例上与正常的 PDB 结构损失联合训练的,并且没有进行预训练。
解释神经网络
为了了解 AlphaFold 如何预测蛋白质结构,我们为网络中的 48 个 Evoformer 块中的每一个训练了一个单独的结构模块,同时保持主网络的所有参数保持不变(补充方法 1.14)。包括我们的回收阶段,这提供了 192 个中间结构的轨迹,每个完整的 Evoformer 块一个,其中每个中间体代表网络对该块最可能结构的信念。在前几个块之后产生的轨迹出奇地平滑,表明 AlphaFold 对结构进行了不断的增量改进,直到它不能再改进为止(见图 4b 的准确度轨迹)。这些轨迹也说明了网络深度的作用。对于非常具有挑战性的蛋白质,如 SARS-CoV-2 Orf8 (T1064),网络搜索并重新排列多层的二级结构元素,然后再确定一个好的结构。对于 LmrP (T1024) 等其他蛋白质,网络会在前几层内找到最终结构。请参阅增刊-视频 1-4 的 CASP14 目标 T1024、T1044、T1064 和 T1091 的结构轨迹显示了一系列蛋白质大小和难度的清晰迭代构建过程。在补充-方法 1.16 和增刊-图12-13,我们解释了 AlphaFold 层产生的注意力图。
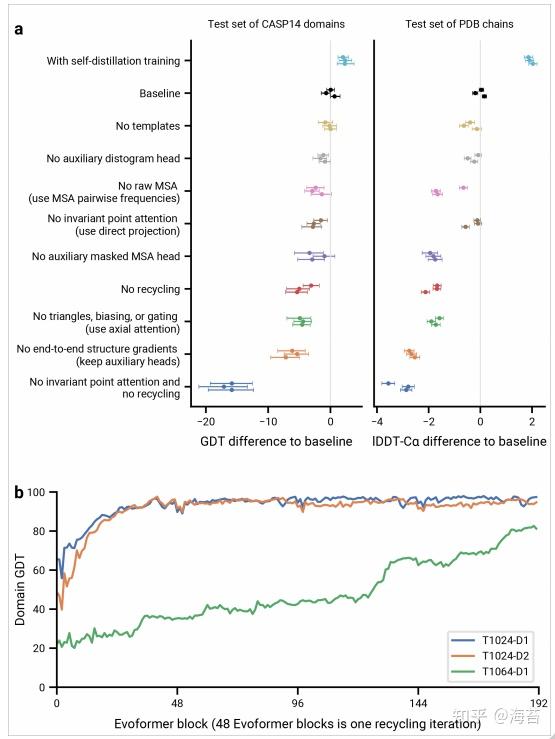
Fig. 4 | Interpreting the neural network
图 4a 包含 AlphaFold 组件的详细消融,表明各种不同的机制有助于 AlphaFold 的准确性。 请参阅增刊-方法 1.13 详细描述了每个消融模型、它们的训练细节、消融结果的扩展讨论以及 MSA 深度对每次消融的影响(补充-图 10)。
MSA 深度和跨链接触
虽然 AlphaFold 在绝大多数沉积的 PDB 结构中具有很高的准确性,但我们注意到仍然存在影响准确性或限制模型适用性的因素。当平均比对深度小于约 30 个序列时,该模型使用多个序列比对,准确度大幅下降(详见图 5a)。我们观察到阈值效应,其中 MSA 深度超过约 100 个序列的改进导致小增益。我们假设需要 MSA 信息在网络的早期阶段粗略地找到正确的结构,但是将该预测细化为高精度模型并不关键取决于 MSA 信息。我们观察到的另一个实质性限制是,与异型接触的数量相比,AlphaFold 对于链内或同型接触很少的蛋白质要弱得多。这通常发生在较大复合物中的桥接结构域,其中蛋白质的形状几乎完全由与复合物中其他链的相互作用产生。相反,AlphaFold 通常能够为同聚体提供高精度预测,即使链基本上交织在一起(例如图 5b)。我们希望 AlphaFold 的想法很容易适用于预测未来系统中的完整异质复合物,并且这将消除具有大量异质接触的蛋白质链的困难。
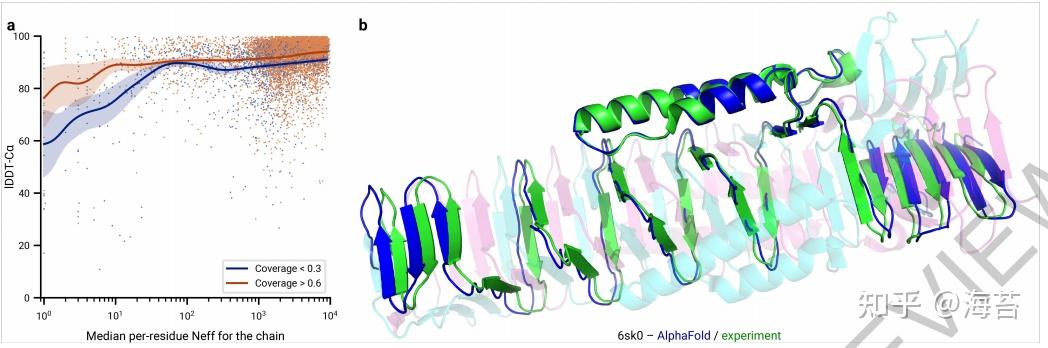
Fig. 5 | Effect of MSA depth and cross-chain contacts.
相关工作
蛋白质结构预测有一个漫长而多样的发展,在许多优秀的评论中得到了广泛的介绍。尽管将神经网络应用于结构预测的历史悠久,但它们最近才开始改进结构预测。这些方法通过将蛋白质结构预测问题处理为将进化耦合的“图像” 转换为蛋白质距离矩阵的“图像”,然后将距离预测集成到启发式系统中,从而有效地利用了计算机视觉系统的快速改进。产生最终的 3-D 坐标预测。最近开发了一些工作来直接预测 3-D 坐标 ,但这些方法的准确性与传统的手工结构预测管道不匹配 。同时,基于注意力的语言处理网络的成功 和最近的计算机视觉激发了对解释蛋白质序列的基于注意力的方法的探索。
讨论
我们在设计 AlphaFold 时采用的方法是生物信息学和物理方法的结合:我们使用物理和几何归纳偏差来构建组件,这些组件可以从 PDB 数据中学习,并最大限度地减少手工制作的特征(例如,AlphaFold 在没有氢的情况下有效地构建氢键债券评分函数)。这使得网络从 PDB 中的有限数据中更有效地学习,但能够应对结构数据的复杂性和多样性。特别是,AlphaFold 能够处理缺失的物理环境,并在具有挑战性的情况下生成准确的模型,例如交织的同源异构体或仅在未知血红素组存在时才会折叠的蛋白质。处理未指定结构条件的能力对于从 PDB 结构中学习至关重要,因为 PDB 代表了结构已被求解的所有条件。一般来说,AlphaFold 被训练产生最有可能作为 PDB 结构的一部分出现的蛋白质结构。在特定化学计量或配体/离子可单独从序列预测的情况下,AlphaFold 可能会产生一种隐式遵守这些约束的结构。
AlphaFold 已经向实验界展示了它的实用性,包括分子置换和解释低温电子显微镜 (cryo-EM) 图 。 此外,由于 AlphaFold 直接输出蛋白质坐标,因此 AlphaFold 会根据蛋白质序列的长度以图形处理单元 (GPU) 分钟到 GPU 小时生成预测(例如,对于 384 个残基,每个模型大约 1 个 GPU 分钟,请参阅方法了解详细信息 )。 这开辟了在蛋白质组规模及其他范围内预测结构的令人兴奋的可能性。
可用基因组测序技术和数据的爆炸式增长彻底改变了生物信息学,但实验结构测定的内在挑战阻止了我们结构知识的类似扩展。 通过开发准确的蛋白质结构预测算法,再加上由实验社区组装的现有大型且精心准备的结构和序列数据库,我们希望加速结构生物信息学的进步,以跟上基因组学革命的步伐。 我们希望 AlphaFold 以及将其技术应用于其他生物物理问题的计算方法将成为现代生物学的重要工具。
在线内容
任何方法、附加参考资料、自然研究报告摘要、源数据、扩展数据、补充信息、致谢、同行评审信息; 作者贡献和竞争利益的详细信息; 数据和代码可用性声明可在 Highly accurate protein structure prediction with AlphaFold – Nature 上获得。
Highly accurate protein structure prediction with AlphaFold – Nature