论文地址: https://www.nature.com/articles/s43588-022-00273-6

中国科大用深度学习实现高实验成功率的蛋白质序列从头设计
中国科学技术大学生命科学与医学部刘海燕教授、陈泉副教授团队与信息科学技术学院李厚强教授团队合作,开发了一种基于深度学习为给定主链结构从头设计氨基酸序列的算法ABACUS-R,在实验验证中,ABACUS-R的设计成功率和设计精度超过了原有统计能量模型ABACUS。相关成果以“Rotamer-Free Protein Sequence Design Based on Deep Learning and Self-Consistency”为题于北京时间2022年7月21日发表于Nature Computational Science。
刘海燕教授、陈泉副教授团队致力于发展数据驱动的蛋白质设计方法,建立并实验验证了利用神经网络能量函数从头设计主链结构的SCUBA模型,以及对给定主链结构设计氨基酸序列的统计能量函数ABACUS。然而,通过优化能量函数来进行序列设计的方法在成功率、计算效率等方面仍有不足。近期有多项研究表明,用深度学习进行氨基酸序列设计能够在天然氨基酸残基类型恢复率等计算指标上超过能量函数方法;但截至目前已正式发表的工作中,对相关方法的实验验证结果远未达到能量函数方法的成功率。该论文报道的ABACUS-R模型,则不仅在计算指标上超过ABACUS,在实验验证中成功率和结构精度也有大幅提高。
用ABACUS-R进行序列设计的方法由两部分组成(图1)。第一部分为预训练的编码器-解码器网络:该网络用Transformer把中心氨基酸残基的化学和空间结构环境映射为隐空间表示向量,再用多层感知机网络将该向量解码为包括中心残基氨基酸类型在内的多种真实特征(图1a)。在方法的第二部分,经用非冗余天然蛋白序列结构数据训练后,ABACUS-R编码器-解码器被用于给定主链结构的全部或部分氨基酸序列从头设计。具体为:从任意初始序列出发,对各个类型待定残基分别应用ABACUS-R编码器-解码器,得到环境依赖的最适宜残基类型,并反复迭代至不同位点的残基类型最大程度自洽(图1b)。
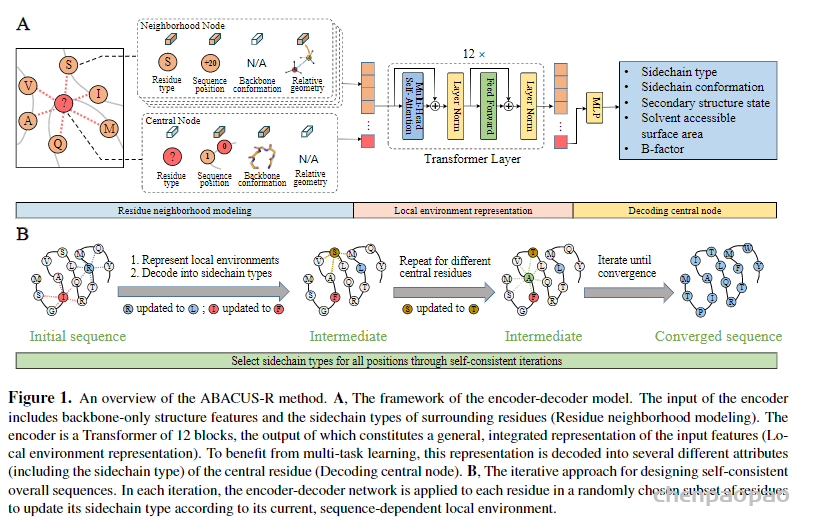
ABACUS-R方法包含两部分:(1)一个encoder-decoder网络被预训练用以推断给定骨架的局部环境时中心残基的侧链类型 (2)用该encoder-decoder网络连续更新每个残基的类型,最终收敛获得自洽(self-consistent)。网络的输入是中心残基与空间上最邻近(Cα间距离)k个残基组成的局部结构。邻近残基的特征包含空间层面的相对位置与取向信息(XSPA)、序列层面的相对位置信息(XRSP)以及邻近残基的残基类型(XAA)。第i个中心残基的特征包含全零的XSPA、被mask的XAA以及骨架上的15个ϕi−2, ψi−2, ωi−2 ⋯ ϕi+2, ψi+2, ωi+2,这些特征组合起来会被映射到与邻近残基特征相同的维度。以上模型输入的信息都是旋转平移不变的。局部结构中的所有残基的特征经过可学习的映射后融合后,得到每个残基总特征En。{En; n = 0, 1, 2, … , k}经过基于 transformer架构的encoder-decoder,预测每个中心残基的类型以及其他辅助任务。
自洽迭代设计的方法是:对序列随机初始化,第一轮随机选择80%的残基通过encoder-decoder并行预测其残基类型,以后每轮随机选择的残基数目逐渐下降。最终的设计结果会逐渐收敛。
在理论验证的基础上,中国科大团队尝试了实验表征用ABACUS-R对3个天然主链结构重新设计的57条序列;其中86%的序列(49条)可溶表达并能折叠为稳定单体;实验解析的5个高分辨晶体结构与目标结构高度一致(主链原子位置均方根位移在1Å以下)(图2)。此外,与以前报道的从头设计蛋白相似,ABACUS-R从头设计的蛋白表现出超高热稳定性,去折叠温度大多可达100℃以上。
作者将PDB中的非冗余结构按照两种不同的方式划分了95%作为训练集、5%作为测试集,第一种划分方式确保测试集的结构不会存在训练集中出现过的CATH拓扑,训练得到的模型为Modeleval;第二种划分方式时随机划分Modelfinal。Modeleval可以用来评估模型能力的无偏向性的表现,而Modelfinal使用了更丰富的数据训练表现应当更好。
表现评估
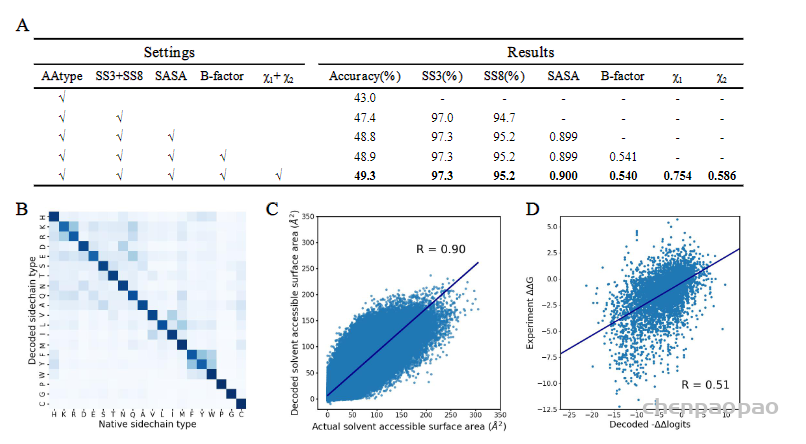
Encoder-decoder的架构可以进行多任务学习,除了训练序列的恢复的任务以外,还可以预测二级结构、SASA、B-factor与侧链扭转角χ1、χ2。多个任务可以增强模型设计序列的能力(图2a),Modeleval与Modelfinal都可以在测试集上最好取得50%左右准确度。在测试集上的结果显示,虽然有些残基类型没有恢复正确,但是模型也学习到了替换为性质相似的残基(图2b)。
相较于ABACUS模型,ABACUS-R序列设计更高的成功率和结构精度进一步增强了数据驱动蛋白质从头设计方法的实用性。ABACUS-R还提供了一种对蛋白质局部结构信息的预训练表示方式,可用于序列设计以外的其他任务。
Decoder网络输出的是每个位置上残基类型的-logP,类似于选择不同残基对应的能量,所以作者将ProTherm数据集中蛋白突变的ΔΔG与模型计算出相应的−ΔΔlogits进行了比较,发现二者有一定的相关性(图2d),说明模型一定程度上学习到了能量。
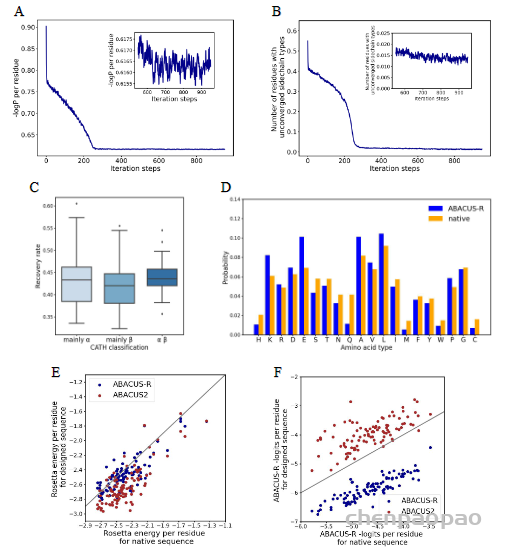
接着,作者验证了模型的自洽性,测试集中100个蛋白属于CATH的三个大类,对其中的每个蛋白从随机序列出发设计10条序列,随着迭代的次数变多,平均-logP会趋于收敛(图3a),同时未收敛的残基比例也会收敛(图3b)。不同CATH类别的骨架上取得的序列恢复率差距不大(图3c)。同一蛋白骨架设计出的序列会有很高的相似性(0.76-0.89)。设计出的序列与天然序列相比,序列的成分高度相似(图3d),Pearson相关系数达到了0.93,但GLU、ALA与LYS出现得更频繁,而Gln、His、Met出现得更少。此外,ABACUS-R设计出的序列与ABACUS设计出的序列相比,平均每个残基的Rosetta打分更低(图3e),而平均的-logP打分却更高(图3f),这意味着ABACUS-R学习到的能量与Rosetta打分函数存在正交的部分。
相较于其他深度学习方法在单个残基恢复任务上的表现,ABACUS-R超过了除DenseCPD外的所有方法(表1),在整条序列重设计任务上ABACUS-R在两个测试集上都取得了最好的表现
实验验证
最后,作者在3种天然骨架(PDB ID: 1r26, 1cy5 and 1ubq)上通过实验验证了ABACUS-R的设计能力。设计的方法有两种:第一种采用迭代自洽的设计方法(生成序列的多样性低),第二种采用迭代时对decoder输出结果进行采样(生成序列的多样性高,但-logP能量也略高)。
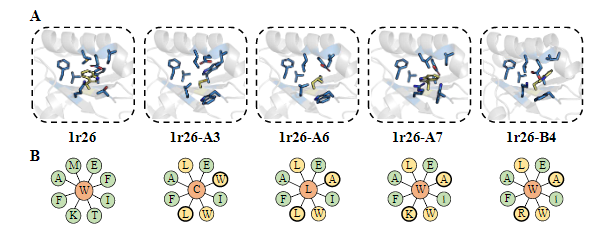
第一种方法设计的27条序列有26条成功表达,体积排阻色谱与1H NMR实验结果显示所有的蛋白都以单体形式存在,示差扫描量热实验显示5条序列有很好的热稳定性( 97~117 ∘C )。最终,1r26的3个设计与1cy5的1个设计成功解出了晶体结构,Cα RMSD位于0.51~0.88 Å,而1ubq的1个设计虽然没有解出结构,但已有的实验结果显示它折叠成了明确的三维结构。
第二种方法对同一骨架设计的序列相似度在58%左右。30条设计的序列中,25条被成功表达,23条能被可溶地纯化。所有设计同样都是单体存在并且折叠成了明确的三维结构,5个设计有很好的热稳定性(85~118 ∘C)。最终,1r26的1个设计被成功解出了晶体结构,Cα RMSD为0.67 Å。相较方法一的自洽设计,方法二设计成功率下降,成功设计的蛋白热稳定性也略微下降,但作者认为可以接受。
最后,作者展示了所有1r26设计晶体结构核心的侧链pack(图4a,b),以及 1cy5设计晶体结构的侧链的极性作用(图4c),说明了ABACUS-R学会了设计侧链的组合以pack好的结构。