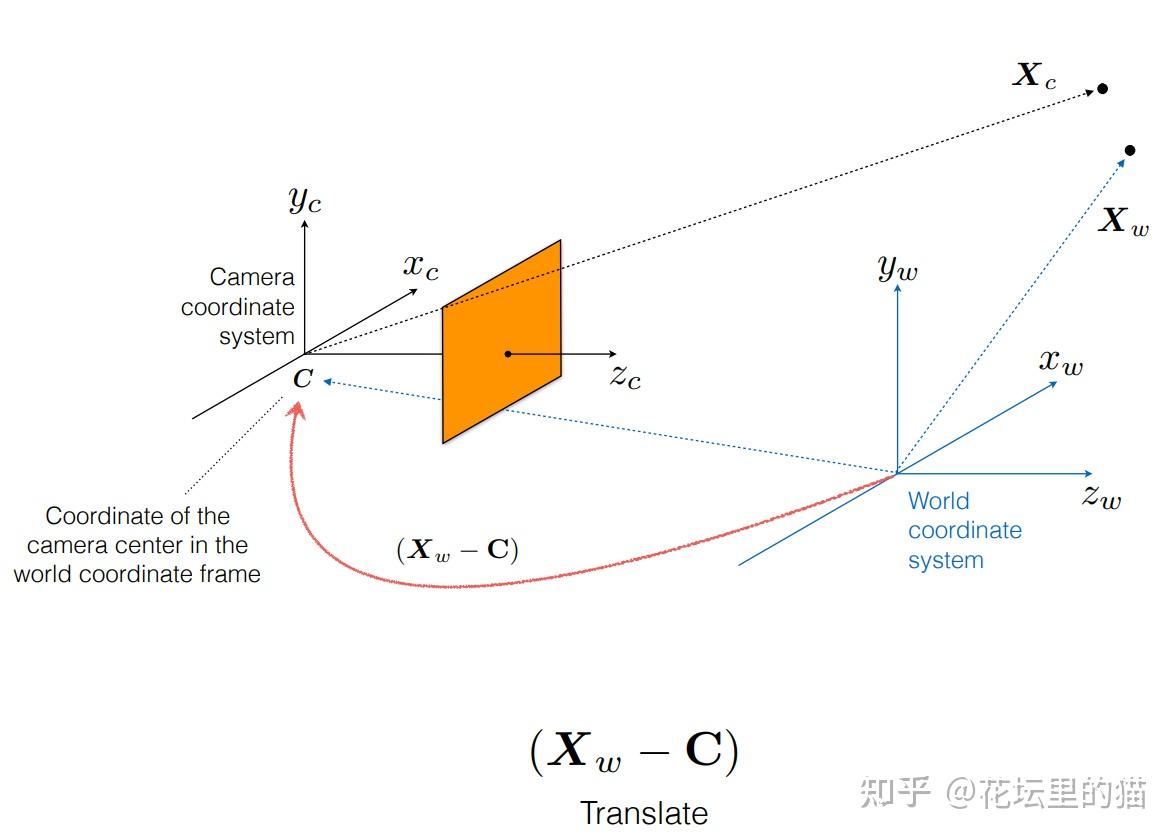
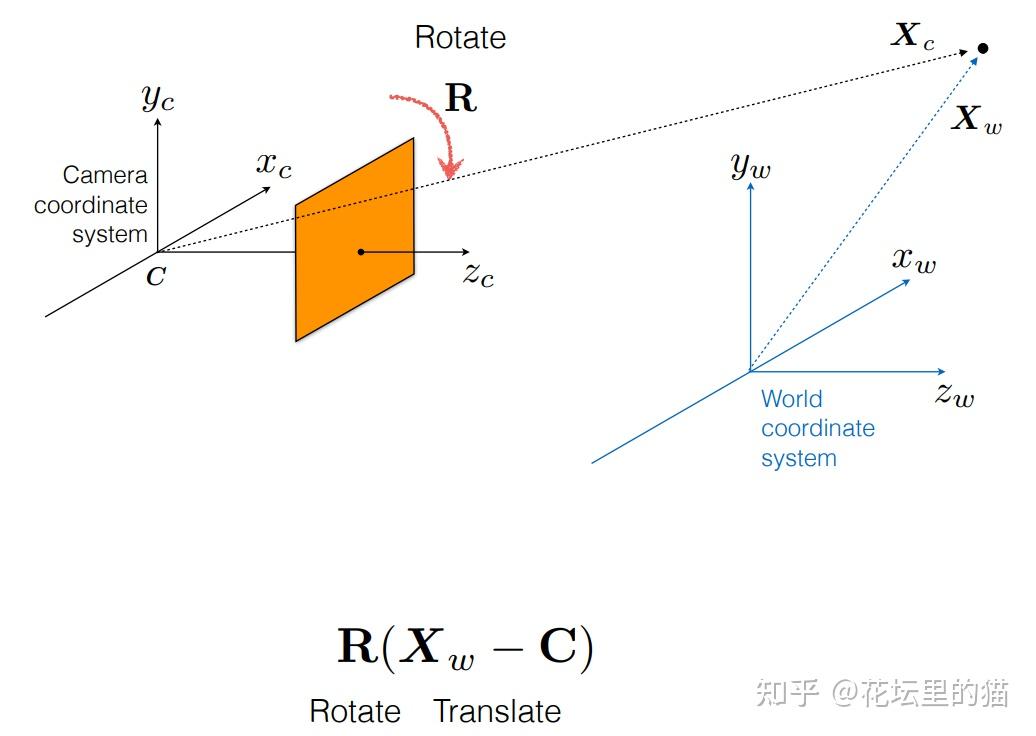
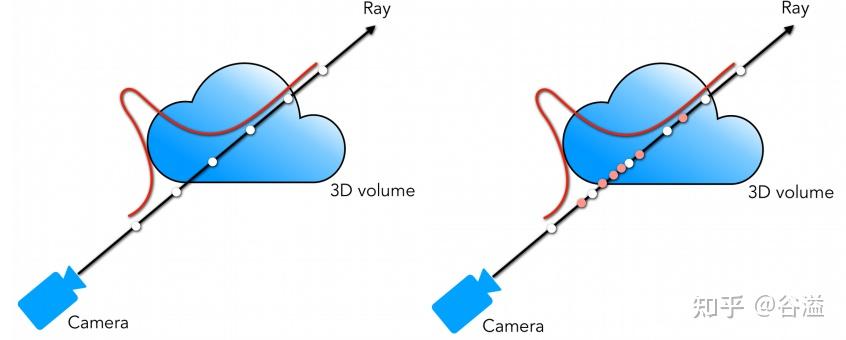
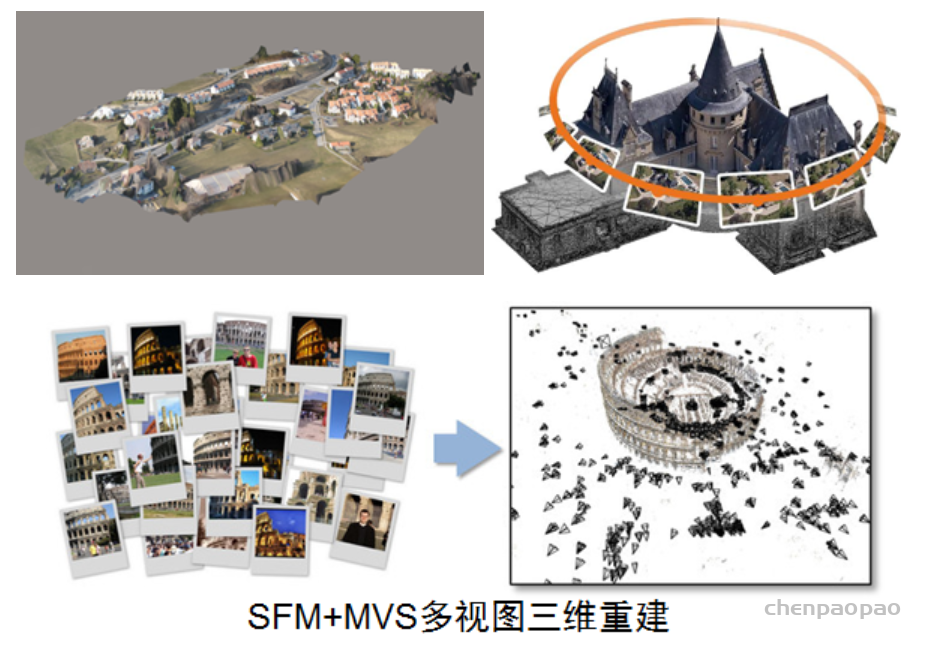
总之, ray tracing就是借由这个概念算出在 image平面上的 2D投影该长什么样子。实际上这边的名词蛮复杂的 (至少对非图学出身的笔者来说),像是 ray tracing、 ray casting、 ray marching什么的。为了避免混淆视听,这边就先不解释过多了。
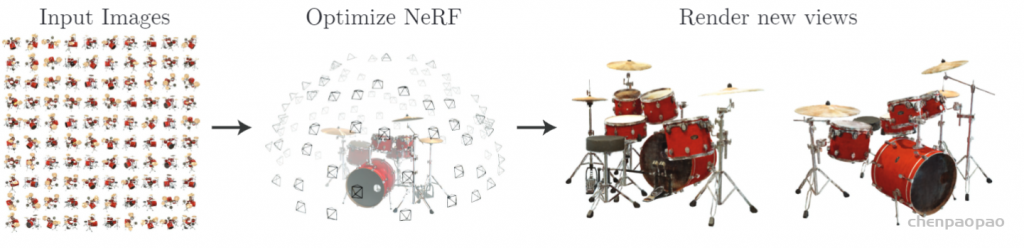
接下来,就开始正式介绍 NeRF的细节。
Neural Radiance Fields (NeRF)-神经辐射场
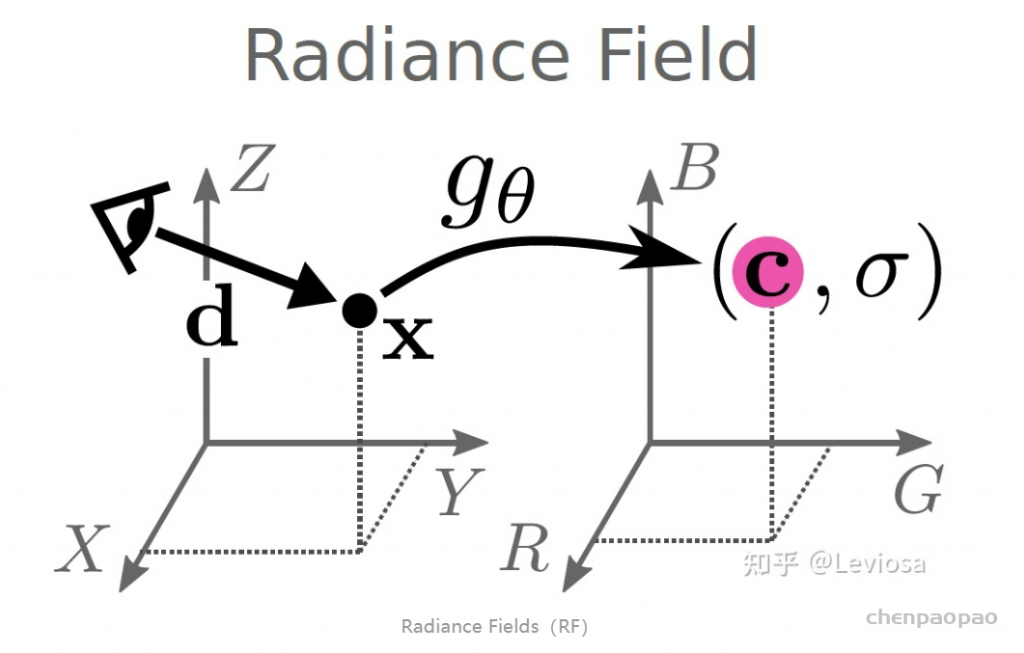
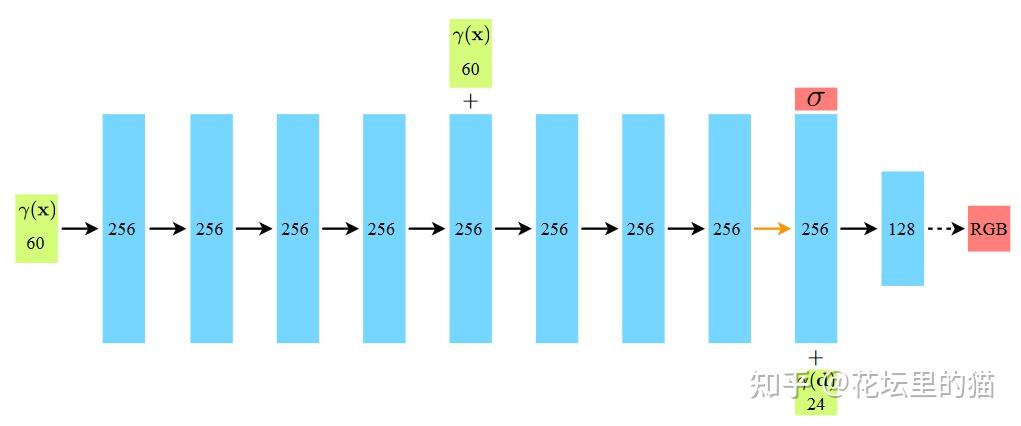
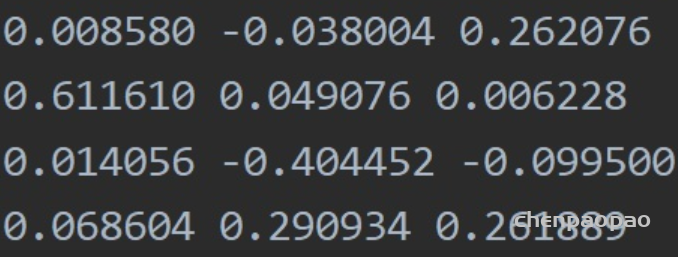
NeRF网路的输入是一组 5D的参数,包含一组 3D的 location X = (x, y, z)跟一组 2D的 view direction (θ, φ),实事上这个 view direction表示为 3D Cartesian unit vector(笛卡尔单位矢量),称为 d。而NeRF网路的输出则是一组 emitted color (如RPG的c=(r, g, b)) 与 volume density σ。
首先由于体素渲染需要沿着光线进行积分,而积分在计算机中是以离散的乘积和进行计算的,那么这里就涉及到在光线上进行点的采样。NeRF在光线的点采样过程中的进行了一些设计。首先为了避免大量的点采样导致的计算量的激增,NeRF设计了coarse to fine的采样策略。在coarse采样阶段,采用了带有扰动的均匀采样方法。第一步在光线的边界之间进行深度空间的均匀采样:
z_steps = torch.linspace(0, 1, N_samples, device=rays.device)
z_vals = near * (1-z_steps) + far * z_steps
在GRAF之后,GIRAFFE实现了composition。在NeRF、GRAF中,一个Neural Radiance Fields表示一个场景,one model per scene。而在GIRAFFE中,一个Neural Radiance Fields只表示一个物体,one object per scene(背景也算一个物体)。这样做的妙处在于可以随意组合不同场景的物体,可以改变同一场景中不同物体间的相对位置,渲染生成更多训练数据中没有的全新图像。
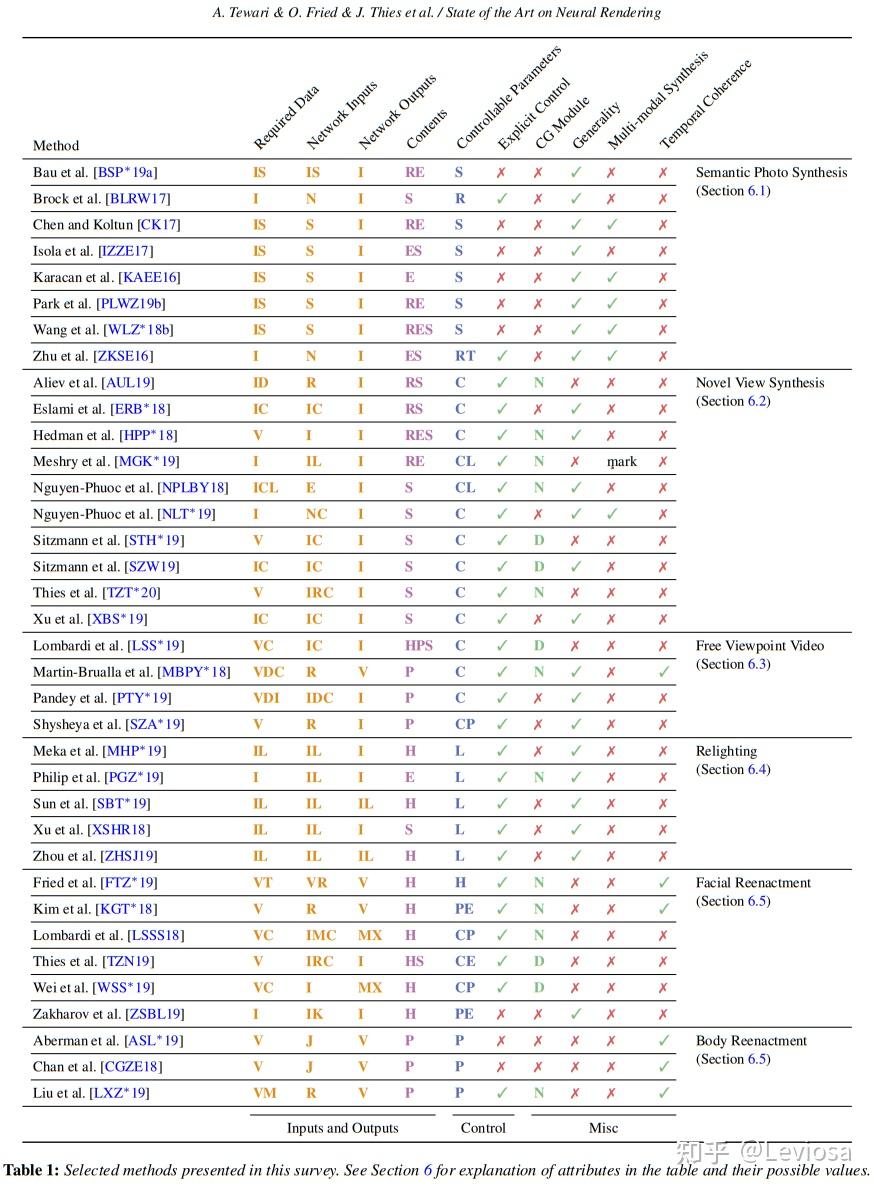
在针对这个更宽泛的概念的综述State of the Art on Neural Rendering中,Neural Rendering的主要研究方向被分为5类,NeRF在其中应属于第2类“Novel View Synthesis”(不过这篇综述早于NeRF发表,表中没有NeRF条目)。
Neural Rendering的5类主要研究方向
表中彩色字母缩写的含义:
在这篇综述中,Neural Rendering被定义为:
Deep image or video generation approaches that enable explicit or implicit control of scene properties such as illumination, camera parameters, pose, geometry, appearance, and semantic structure.
融入了 Prompt 的模式大致可以归纳成 “Pre-train, Prompt, and Predict”,在该模式中,下游任务被重新调整成类似预训练任务的形式。例如,通常的预训练任务有 MLM(Masked Language Model),在文本情感分类任务中,对于 “I love this movie” 这句输入,可以在后面加上 Prompt:”the movie is ___”,组成如下这样一句话:
Prompt 在 PLM debias 方面的应用。由于 PLM 在预训练过程中见过了大量的人类世界的自然语言,所以很自然地会受到一些影响。举一个简单的例子,比如说训练语料中有非常多 “The capital of China is Beijing”,导致模型每次看到 “capital” 的时候都会预测出 “Beijing”,而不是去分析到底是哪个国家的首都。在应用的过程中,Prompt 还暴露了 PLM 学习到的很多其它 bias,比如种族歧视、性别对立等。这也许会是一个值得研究的方向
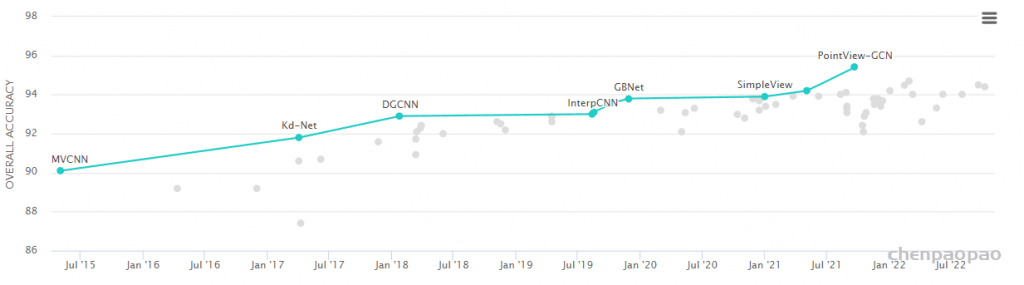
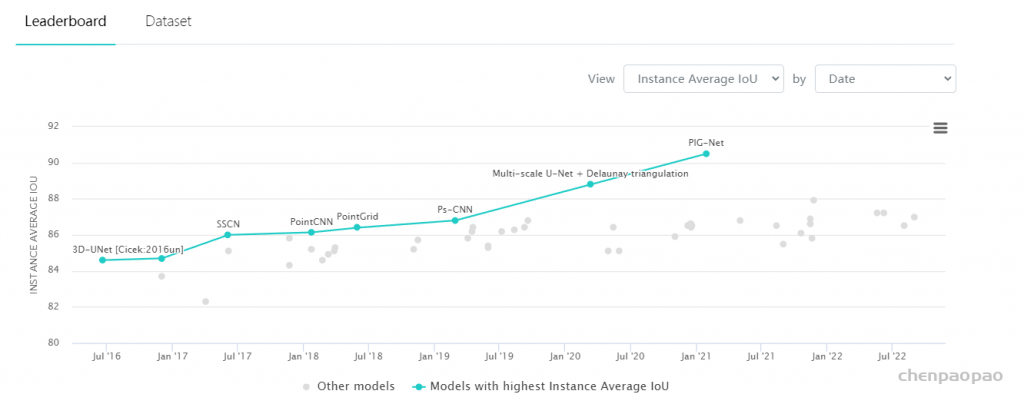
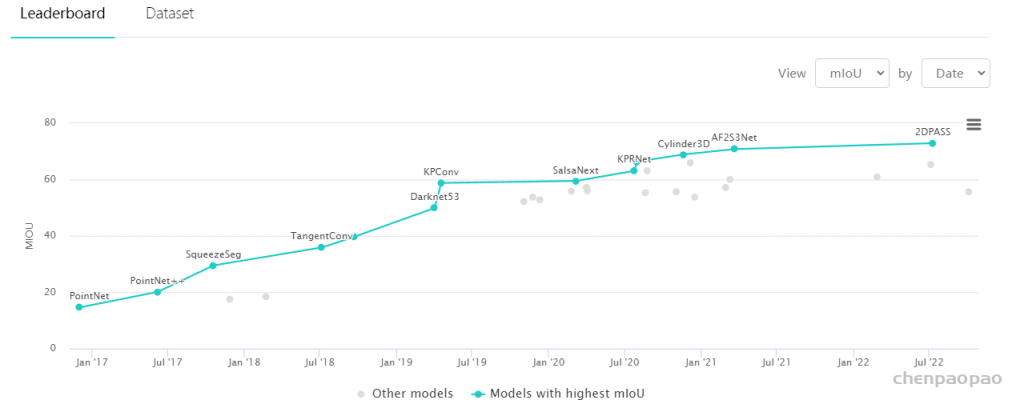
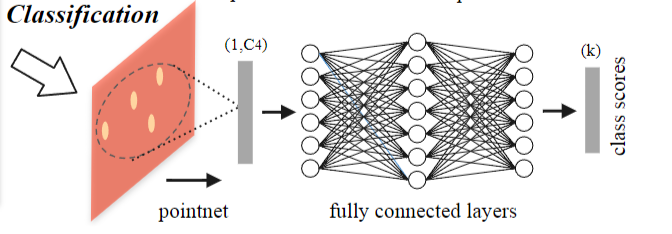
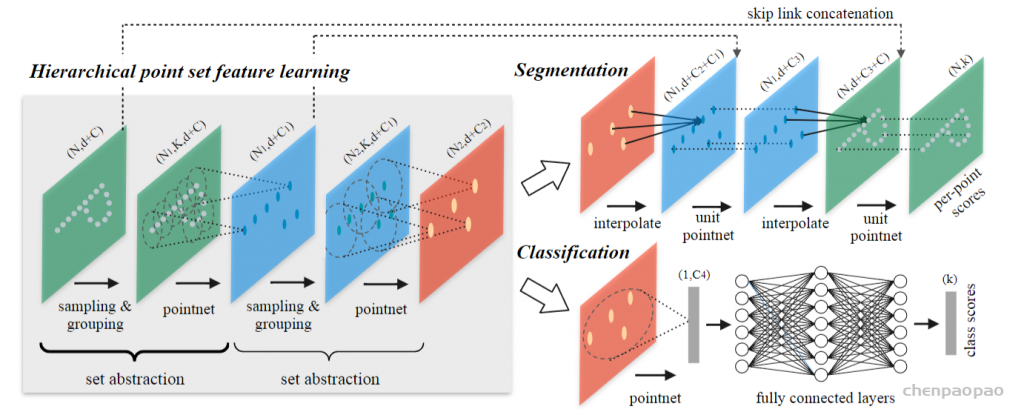
3D Point Cloud Classification on ModelNet40 3D Point Cloud Classification on ModelNet40 3D Part Segmentation on ShapeNet-Part3D Semantic Segmentation on SemanticKITTI
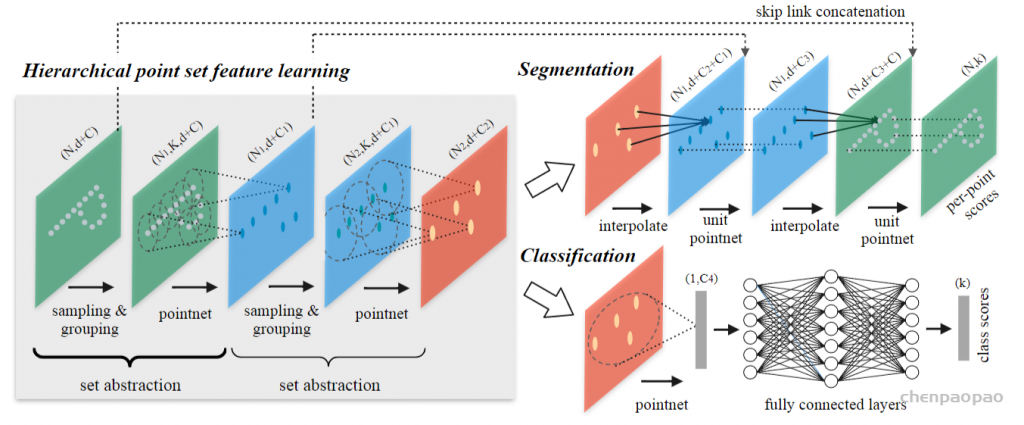
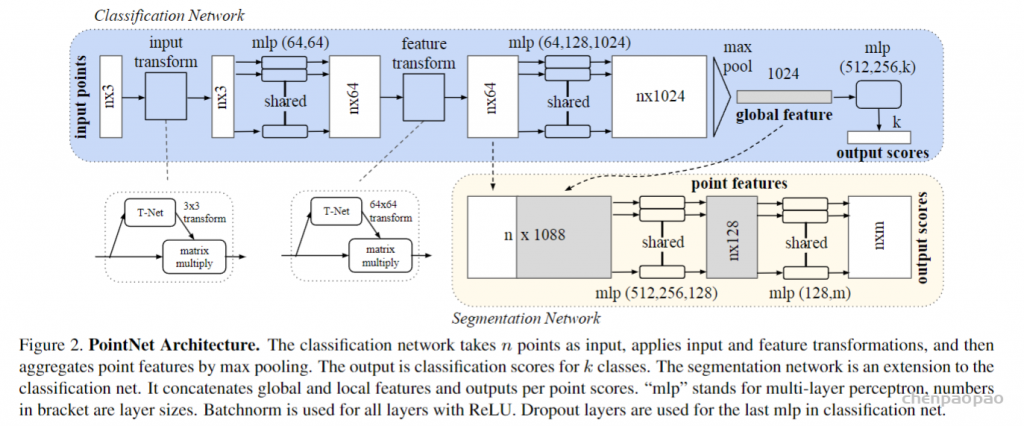
def pointnet_sa_module(xyz, points, npoint, radius, nsample, mlp, mlp2, group_all, is_training, bn_decay, scope, bn=True, pooling='max', knn=False, use_xyz=True, use_nchw=False):
''' PointNet Set Abstraction (SA) Module
Input:
xyz: (batch_size, ndataset, 3) TF tensor
points: (batch_size, ndataset, channel) TF tensor
npoint: int32 -- #points sampled in farthest point sampling
radius: float32 -- search radius in local region
nsample: int32 -- how many points in each local region
mlp: list of int32 -- output size for MLP on each point
mlp2: list of int32 -- output size for MLP on each region
group_all: bool -- group all points into one PC if set true, OVERRIDE
npoint, radius and nsample settings
use_xyz: bool, if True concat XYZ with local point features, otherwise just use point features
use_nchw: bool, if True, use NCHW data format for conv2d, which is usually faster than NHWC format
Return:
new_xyz: (batch_size, npoint, 3) TF tensor
new_points: (batch_size, npoint, mlp[-1] or mlp2[-1]) TF tensor
idx: (batch_size, npoint, nsample) int32 -- indices for local regions
'''
data_format = 'NCHW' if use_nchw else 'NHWC'
with tf.variable_scope(scope) as sc:
# Sample and Grouping
if group_all:
nsample = xyz.get_shape()[1].value
new_xyz, new_points, idx, grouped_xyz = sample_and_group_all(xyz, points, use_xyz)
else:
new_xyz, new_points, idx, grouped_xyz = sample_and_group(npoint, radius, nsample, xyz, points, knn, use_xyz)
# Point Feature Embedding
if use_nchw: new_points = tf.transpose(new_points, [0,3,1,2])
for i, num_out_channel in enumerate(mlp):
new_points = tf_util.conv2d(new_points, num_out_channel, [1,1],
padding='VALID', stride=[1,1],
bn=bn, is_training=is_training,
scope='conv%d'%(i), bn_decay=bn_decay,
data_format=data_format)
if use_nchw: new_points = tf.transpose(new_points, [0,2,3,1])
# Pooling in Local Regions
if pooling=='max':
new_points = tf.reduce_max(new_points, axis=[2], keep_dims=True, name='maxpool')
elif pooling=='avg':
new_points = tf.reduce_mean(new_points, axis=[2], keep_dims=True, name='avgpool')
elif pooling=='weighted_avg':
with tf.variable_scope('weighted_avg'):
dists = tf.norm(grouped_xyz,axis=-1,ord=2,keep_dims=True)
exp_dists = tf.exp(-dists * 5)
weights = exp_dists/tf.reduce_sum(exp_dists,axis=2,keep_dims=True) # (batch_size, npoint, nsample, 1)
new_points *= weights # (batch_size, npoint, nsample, mlp[-1])
new_points = tf.reduce_sum(new_points, axis=2, keep_dims=True)
elif pooling=='max_and_avg':
max_points = tf.reduce_max(new_points, axis=[2], keep_dims=True, name='maxpool')
avg_points = tf.reduce_mean(new_points, axis=[2], keep_dims=True, name='avgpool')
new_points = tf.concat([avg_points, max_points], axis=-1)
# [Optional] Further Processing
if mlp2 is not None:
if use_nchw: new_points = tf.transpose(new_points, [0,3,1,2])
for i, num_out_channel in enumerate(mlp2):
new_points = tf_util.conv2d(new_points, num_out_channel, [1,1],
padding='VALID', stride=[1,1],
bn=bn, is_training=is_training,
scope='conv_post_%d'%(i), bn_decay=bn_decay,
data_format=data_format)
if use_nchw: new_points = tf.transpose(new_points, [0,2,3,1])
new_points = tf.squeeze(new_points, [2]) # (batch_size, npoints, mlp2[-1])
return new_xyz, new_points, idx
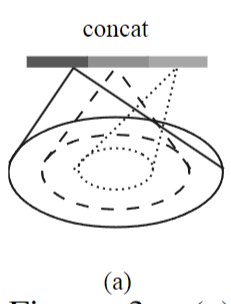
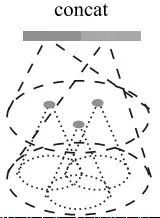
还有个问题:query ball point如何保证对于每个局部邻域,采样点的数量都是一样的呢? 事实上,如果query ball的点数量大于规模 K ,那么直接取前 K 个作为局部邻域;如果小于,那么直接对某个点重采样,凑够规模 K
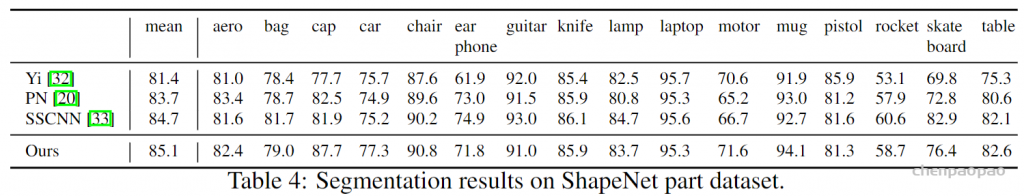
KNN和query ball的区别:(摘自原文)Compared with kNN, ball query’s local neighborhood guarantees a fixed region scale thus making local region feature more generalizable across space, which is preferred for tasks requiring local pattern recognition (e.g. semantic point labeling).也就是query ball更加适合于应用在局部/细节识别的应用上,比如局部分割。
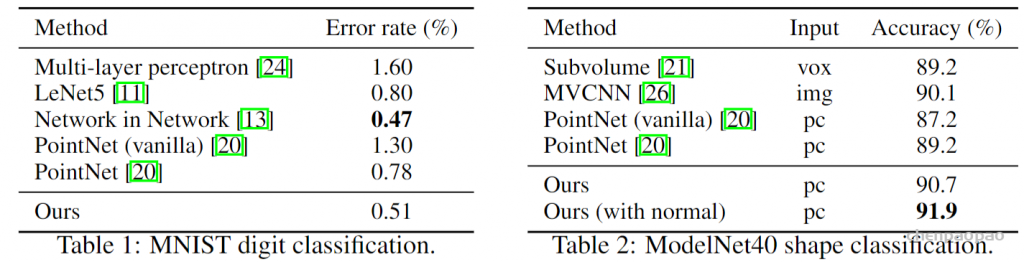
MNIST: Images of handwritten digits with 60k training and 10k testing samples.(用于分类)
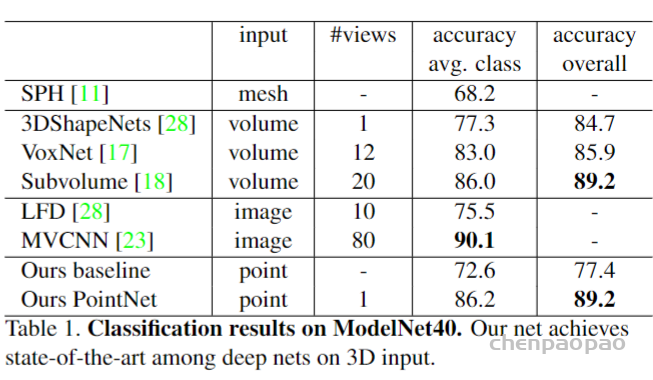
ModelNet40: CAD models of 40 categories (mostly man-made). We use the official split with 9,843 shapes for training and 2,468 for testing. (用于分类)
SHREC15: 1200 shapes from 50 categories. Each category contains 24 shapes which are mostly organic ones with various poses such as horses, cats, etc. We use five fold cross validation to acquire classification accuracy on this dataset. (用于分类)
ScanNet: 1513 scanned and reconstructed indoor scenes. We follow the experiment setting in [5] and use 1201 scenes for training, 312 scenes for test. (用于分割)