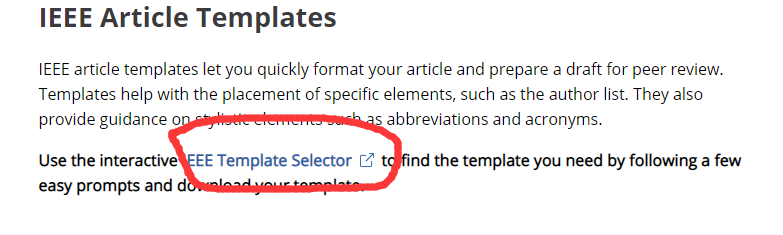
MLP-Mixer是ViT团队的另一个纯MLP架构的尝试。如果MLP-Mixer重新引领CV领域主流架构的话,那么CV领域主流架构的演变过程就是MLP->CNN->Transformer->MLP? 要回到最初的起点了吗???( Transformer移除了注意力以后就剩MLP了)
这篇论文提出了一种”纯“MLP结构的视觉架构。
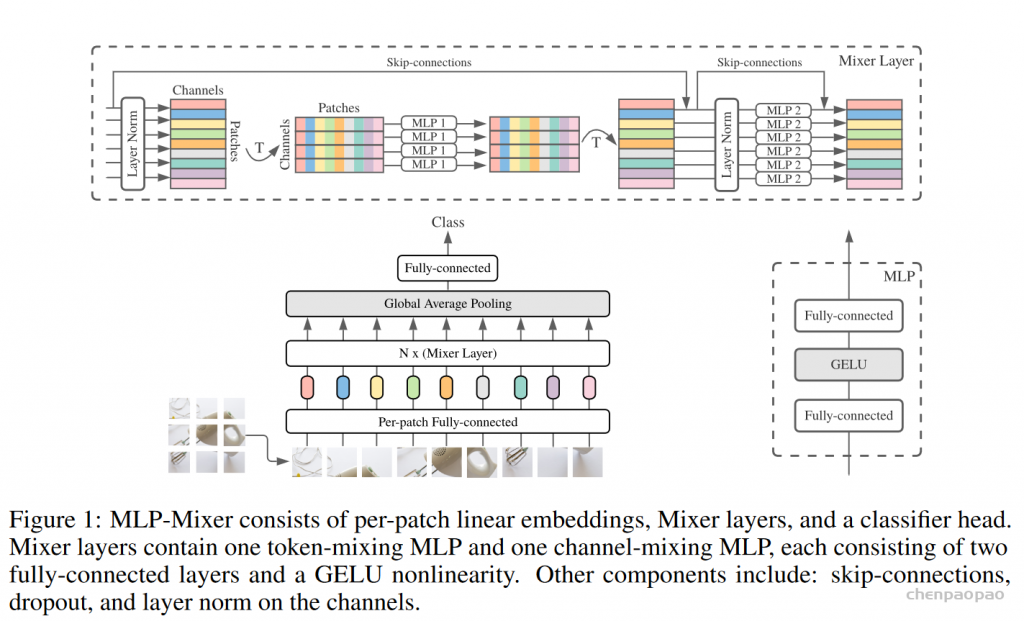
先将输入图片拆分成patches,然后通过Per-patch Fully-connected将每个patch转换成feature embedding,然后送入N个Mixer Layer,最后通过Fully-connected进行分类。
Mixer分为channel-mixing MLP和token-mixing MLP两类。channel-mixing MLP允许不同通道之间进行交流;token-mixing MLP允许不同空间位置(tokens)进行交流。这两种类型的layer是交替堆叠的,方便支持两个输入维度的交流。每个MLP由两层fully-connected和一个GELU构成。
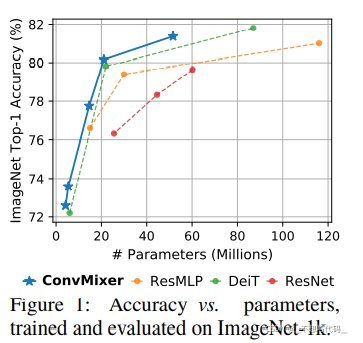
从上图我们可以看出,MLP -Mixer 首先使用图片分成很多个小正方形的patch,每个patch的大小定义为patch_size。论文中实现这一步骤使用的是前面提到的卷积,卷积核的大小和步长均patch_size。论文中给的参数,也是2的幂。
网络不再使用传统的RELU激活函数,而是使用了GELU激活函数。
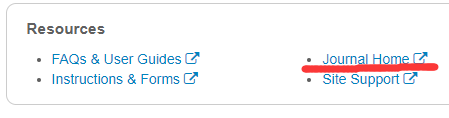
将图片分成小块后,在将它转换为一维结构。如图:
然后将每一个patch进行转换,如下图所示:
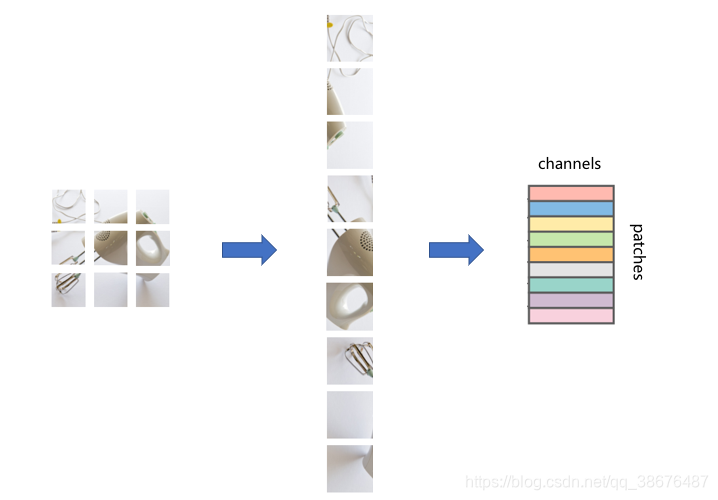
通过这样一种方式呢,就将一张图片转换为了一个大矩阵,就可以输入到Mixer Layer 中进行计算。

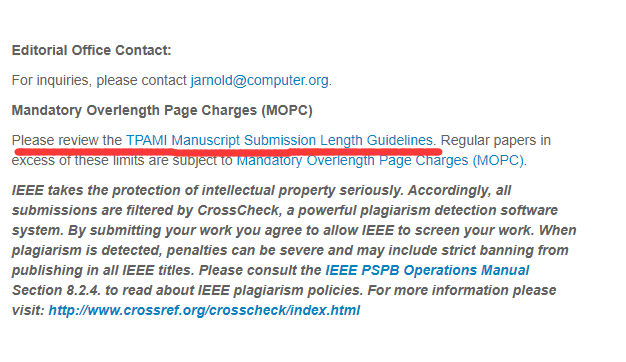
MLP 是两个全连接层的感知机,W1,W2,对应token_mixer中两个全连接的权重,W3,W4则表示channel_mixer两个全连接的权重。σ表示GELU激活函数。那么公示就很简单了,输入X经过Layer Normalize,再乘以W1,再经过激活函数后乘以W2,再加上X。第二个公式也是相同的计算过程。
将前面通过编码得到的矩阵经过Layer Norm 在将矩阵进行旋转(T 表示旋转)连接MLP1,MLP1 就是文章token_mixer 用来寻找像素与像素之间的关系,其中,MLP1中的权值共享。计算完之后,再将矩阵旋转回来,通过Layer Norm 后再接一个channel_mixer 用于寻找通道与通道之间的关系。其中MixerLayer 还启用了ResNet中的跨连结构,跨连结构的作用可以参考[ResNet原理讲解和复现],看到这里,是不是感觉它跟卷积的原理很类似。
从上图可以看出Mixer Layer的输入维度和输出维度相同,并且通过MLP的方式来寻找图片像素与像素,通道与通道的关系。
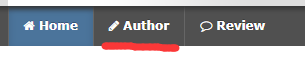
这就是MLP-MIXER的网络结构了
实现的难点在于,矩阵旋转,我们使用einops中的Rearrange实现矩阵旋转
使用Rearrange 实现旋转
Rearrange(‘b n d -> b d n’) #这里是[batch_size, num_patch, dim] -> [batch_size, dim, num_patch]
#定义多层感知机
import torch
import numpy as np
from torch import nn
from einops.layers.torch import Rearrange
from torchsummary import summary
import torch.nn.functional as F
class FeedForward(nn.Module):
def __init__(self,dim,hidden_dim,dropout=0.):
super().__init__()
self.net=nn.Sequential(
#由此可以看出 FeedForward 的输入和输出维度是一致的
nn.Linear(dim,hidden_dim),
#激活函数
nn.GELU(),
#防止过拟合
nn.Dropout(dropout),
#重复上述过程
nn.Linear(hidden_dim,dim),
nn.Dropout(dropout)
)
def forward(self,x):
x=self.net(x)
return x
class MixerBlock(nn.Module):
def __init__(self,dim,num_patch,token_dim,channel_dim,dropout=0.):
super().__init__()
self.token_mixer=nn.Sequential(
nn.LayerNorm(dim),
Rearrange('b n d -> b d n'), #这里是[batch_size, num_patch, dim] -> [batch_size, dim, num_patch]
FeedForward(num_patch,token_dim,dropout),
Rearrange('b d n -> b n d') #[batch_size, dim, num_patch] -> [batch_size, num_patch, dim]
)
self.channel_mixer=nn.Sequential(
nn.LayerNorm(dim),
FeedForward(dim,channel_dim,dropout)
)
def forward(self,x):
x=x+self.token_mixer(x)
x=x+self.channel_mixer(x)
return x
class MLPMixer(nn.Module):
def __init__(self,in_channels,dim,num_classes,patch_size,image_size,depth,token_dim,channel_dim,dropout=0.):
super().__init__()
assert image_size%patch_size==0
self.num_patches=(image_size//patch_size)**2
#embedding 操作,用卷积来分成一小块一小块的
self.to_embedding=nn.Sequential(nn.Conv2d(in_channels=in_channels,out_channels=dim,kernel_size=patch_size,stride=patch_size),
Rearrange('b c h w -> b (h w) c')
)
#经过Mixer Layer 的次数
self.mixer_blocks=nn.ModuleList([])
for _ in range(depth):
self.mixer_blocks.append(MixerBlock(dim,self.num_patches,token_dim,channel_dim,dropout))
self.layer_normal=nn.LayerNorm(dim)
self.mlp_head=nn.Sequential(
nn.Linear(dim,num_classes)
)
def forward(self,x):
x=self.to_embedding(x)
for mixer_block in self.mixer_blocks:
x=mixer_block(x)
x=self.layer_normal(x)
x=x.mean(dim=1)
x=self.mlp_head(x)
return x
MLP-Mixer用Mixer的MLP来替代ViT的Transformer,减少了特征提取的自由度,并且巧妙的可以交替进行patch间信息交流和patch内信息交流,从结果上来看,纯MLP貌似也是可行的,而且省去了Transformer复杂的结构,变的更加简洁,有点期待后续ViT和MLP-Mixer如何针锋相对的,感觉大组就是东挖一个西挖一个的,又把尘封多年的MLP给挖出来了