任务:
1、安装学习yutobe上面的软件
https://www.youtube.com/watch?v=Sux91FJ3Xe8&t=629s
2、跑通论文代码
https://www.youtube.com/watch?v=Sux91FJ3Xe8&t=629s
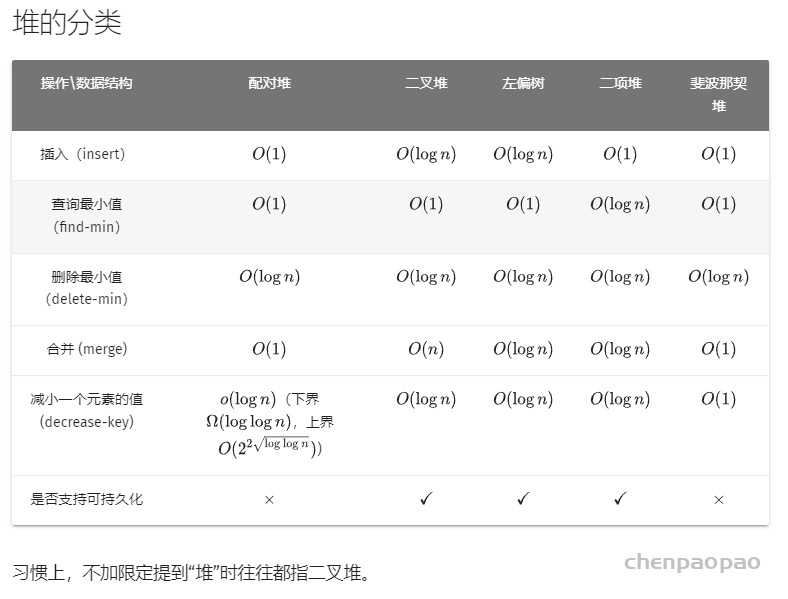
Pymol简介
Pymol是一款操作简单,功能强大的分子以及蛋白的可视化软件,由薛定谔公司研发,科研人员可以从官网申请最新教育版本,同时pymo的开源版(https://github.com/schrodinger/pymol-open-source ),可以直接从网站上下载,但是版本较老。所以,根据需求选择版本进行下载。 说明: https://cloud.tencent.com/developer/article/1785088
Pymol入门教程:
http://pymol.chenzhaoqiang.com/intro/startManual.html
分子对接教程
1、
https://cloud.tencent.com/developer/inventory/15332
2、https://www.bilibili.com/video/av466685164?from=search&seid=9870338638011316620&spm_id_from=333.337.0.0
vina只负责对接,mgltool负责提供蛋白质分子和配体分子。
先用MGL 生成vina需要的pdbqt文件
MGL tools的作用就是生成pdbqt文件
1、:打开MGL tools,打开受体蛋白的pdb :file-》read molecule
2、蛋白质的pdb(数据库):
关于蛋白质结构的PDB文件,做分子对接,估计大家都知道PDB这个蛋白质数据库 啦。这里简单的介绍一下。
蛋白质的三级结构是指整条多肽链的三维空间结构,也就是包括碳骨架和侧链在内的所有原子的空间排列。第一个蛋白质的三维空间结构于 1958 年用 X-射线衍射法(X-ray Crystallography)测定。这种方法目前仍然是获取蛋白质三级结构的主要方法。PDB 数据库中绝大多数蛋白质结构都是用这种方法测定的。另一个测定蛋白质三维空间结构的方法是核磁共振法(Nuclear Magnetic Resonance, NMR)。无法结晶的蛋白质,可以利用核磁共振法在液体环境中进行结构测定。但是核磁共振法只能用于质量小于 70 千道尔顿的分子,大约对应 200 个氨基酸的长度。除此之外,还有一些不太常用的方法也可以测定分子的三维空间结构,比如冷冻电子显微镜技术(Cyro-Electron Microscopy)。无论用什么方法测定的空间结构,都要提交到 PDB 数据库。所以我们获取蛋白质三级结构最直接的办法就是去PDB 搜索(http://www.rcsb.org/ )。 从PDB首页的搜索条里,可以通过搜索PDB ID、分子名称、作者姓名等关键词来查找蛋白质三级结构。此外,利用高级搜索工具,可以通过序列相似性搜索获得与输入序列在序列水平上相似的蛋白质的三级结构。搜索方法选 BLAST,输入序列,点击“Result Count”。这里不详细介绍,因为我们做分子对接,通常蛋白名称是已知的。我们重点介绍怎么选择合适的蛋白结构文件。 比如我们搜索PI3K这个蛋白,直接在搜索栏搜索,结果是有很多的。可以看到有393个结构信息。首先我们可以通过左边的栏进行筛选,比如物种信息,我们选择人。当然,结果的显示排序可通过结果上面的选项卡进行选择不同的排序方式。我们筛选合适的蛋白结构,常用Score这个选项.我们选择分辨率较好的在前。这里的0.9Å,Å是光波长度和分子直径的常用计量单位,值越小,分辨率越高,结构越准确。页面往下拉,可以看见这个值越来越大,我们优先选择值小的。我们可以从页面里面看见一下基本信息,比如方法,物种以及被解析的时间等。这里5GJI这个结构获取的方法就是X-RAY。我们点击这个蛋白,进入后可以看见详细的信息。然后我们还要看这个蛋白的描述是不是我们想要的蛋白,从这里面感觉看起来比较费劲。这里我们借助uniprot这个数据库来选择是比较方便的。这里简单介绍一下这个数据库,可能有的同学是第一次知道。翻了多年前的笔记,粘贴在下面。 UniProt 数据库有三个层次。
第一层叫 UniParc,收录了所有 UniProt 数据库子库中的蛋白质序列,量大,粗糙。
第二层是 UniRef,他归纳了 UniProt 几个主要数据库并且是将重复序列去除后的数据库。
第三层是 UniProtKB,他有详细注释并与其他数据库有链接,分为 UniProtKB 下的 Swiss-Prot和 UniProtKB 下的 TrEMBL 数据库。
关系稍有点复杂,但实际上我们最常用的就是 UniProtKB下的 Swiss-Prot 数据库。
从 UniProt 数据库查看一条蛋白质序列(http://www.uniprot.org/ )。在UniProt数据库的首页上有一个关于 UniProtKB 数据库的统计表。可以看到,TrEMBL 数据库里存储的序列数量远远大于 Swiss-Prot 中的。统计表里清楚的写着:TrEMBL 是自动注释的,没有经过检查,而 Swiss-Prot 是人工注释的,并且经过检查。
然后点击下载文件就可以直接下载PDB格式的蛋白结构文件。下载的PDB文件可以用pymol或者VMD观察结构。能够实现蛋白质三维结构可视化的软件非常多。比专业级的PyMOL(https://pymol.org/2/ )。这个软件已经被世界上著名的生物医药软件公司“薛定谔公司(Schrödinger)”收购。这种专业级的可视化软件不仅能够做出非常漂亮的图片,它还有强大的插件支持各种各样的蛋白质结构分析,这款软件需要购买,如果你发表的文章里提到某些内容是使用PyMOL制作的,而文章中所有作者和作者单位都没有PyMOL的购买记录的话,你可能会面临薛定谔公司的追责。 如果要对接的蛋白没有结构,我们又要对接,那就只能是自己通过软件预测了。蛋白质结构预测的方法有从头计算法,同源建模法,穿线法和综合法。常用的是同源建模法,SWISS-MODEL(www.swissmodel.expasy.org )就是一款用同源建模法预测蛋白质三级结构的全自动软件,这里不详细介绍了,预测的模型还要涉及模型好坏的评价,后续有时间,再介绍蛋白质三级结构的预测。
接下来我们打开AutoDockTools(ADT),打开我们前面保存的文件1E8Y_PYMOL.pdb
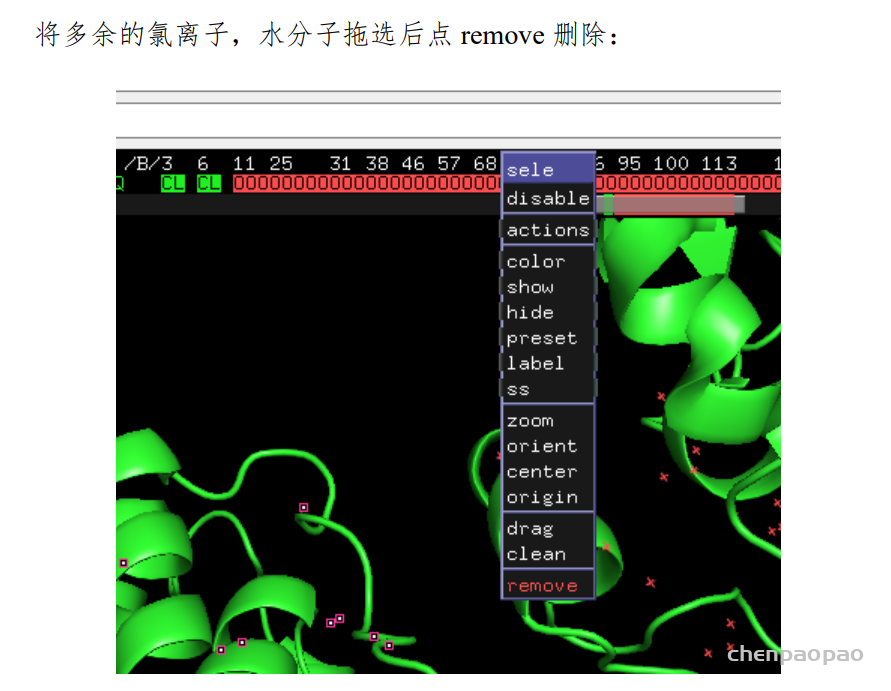
删除水分子和其他配体,常规操作不用解释
然后计算电荷和添加原子类型
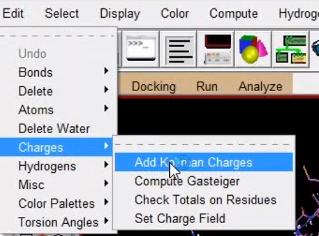
Edit–Charges–Compute Gasteiger
Edit–Atoms–Assign AD4 type
就可以导出成pbdqt格式的文件了
然后右键吧蛋白删除掉,导入配体小分子,随便从ZINC下了一个
ZINC(http://zinc.docking.org/ ) 还有一个数据库能下载mol2格式的文件。ZINC),这里就不介绍了,你要是能从上面的数据库下载到你配体小分子的mol2格式文件,就直接用,如果不能,那就是去PubChem数据库(https://pubchem.ncbi.nlm.nih.gov/ )下载sdf文件,然后进行转换,这也是我这里要介绍的。
Ligand-input-open
分子对接教程
1 分子对接的工作环境
2 准备受体、配体的 pdbqt 文件
2.1.2 关于 pdb 文件的格式
该格式省略了一切氢原子,包括游离的水分子都只保留了一个 O,所以后续需要加氢操作。该文件的格式较复杂,最好使用成熟的软件进行编辑,而不上自行编辑。该文件中的氨基酸残基的原子全部记录在 ATOM 行中,后面的每一项分别为原子序号、原子名称(第一个字母为原子的元素符号,第二个字母为远近标识符 A、B、G、D、E、Z、H 分别对应有机化合物命名系统中的 α、β、γ、δ、ε、ζ、η)、残基名称、链编号、残基序号、原子坐标等
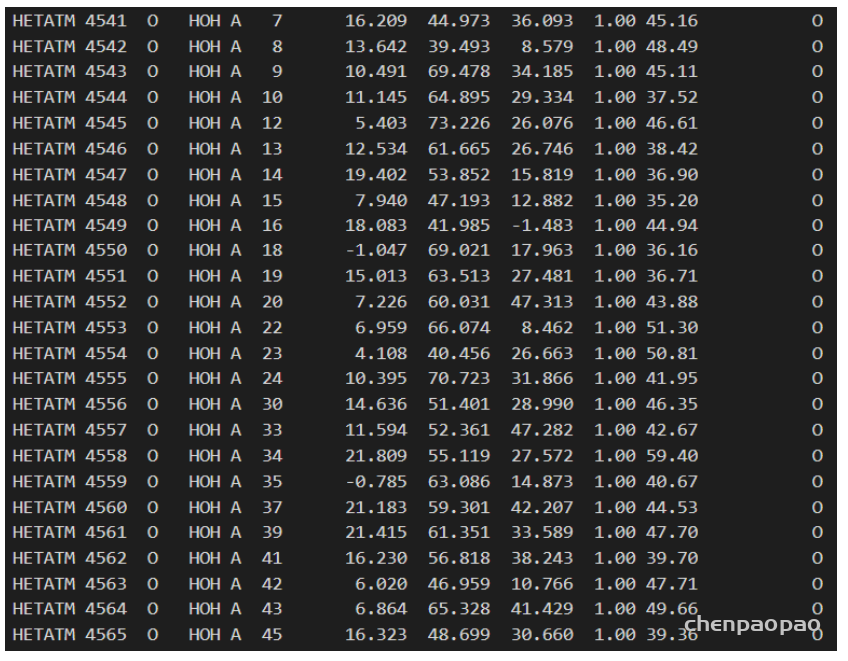
而离子、水分子以及结合在蛋白中的配体(如抑制剂等)等非蛋白质的部分记录在 HETNAM(非标准残基的名称)中:
同样的依次是原子编号、原子名称、基团名称(如水这里起名为HOH)、链编号(但是它本身不依附于哪条链)、原子编号、坐标等。
2.1.3 使用 PyMol 命令进行选择和删除
2.1.1 保存
2.1.1 保存
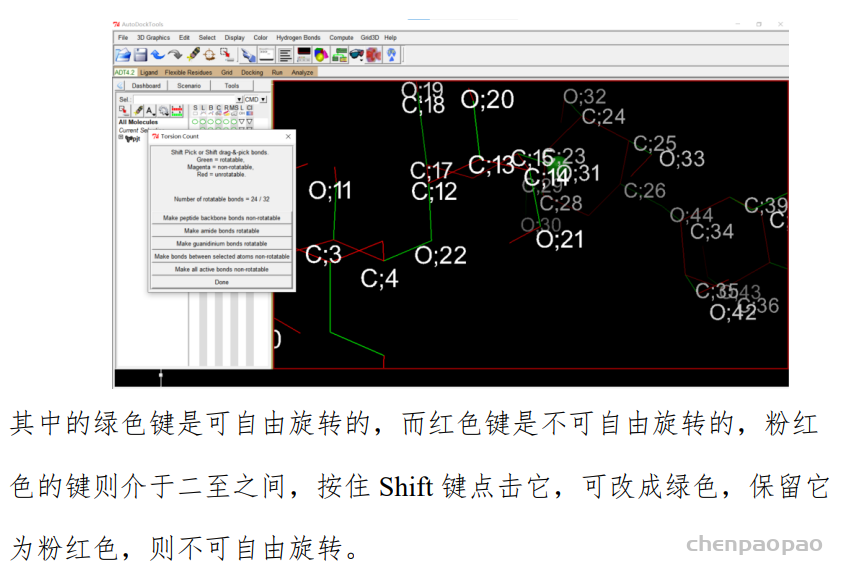
设置好后点 Done,然后点击 Ligand-Output-Save as PDBQT 即可
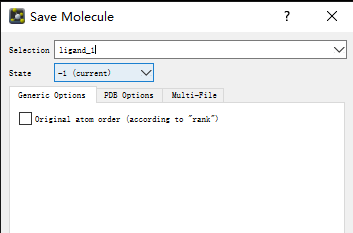
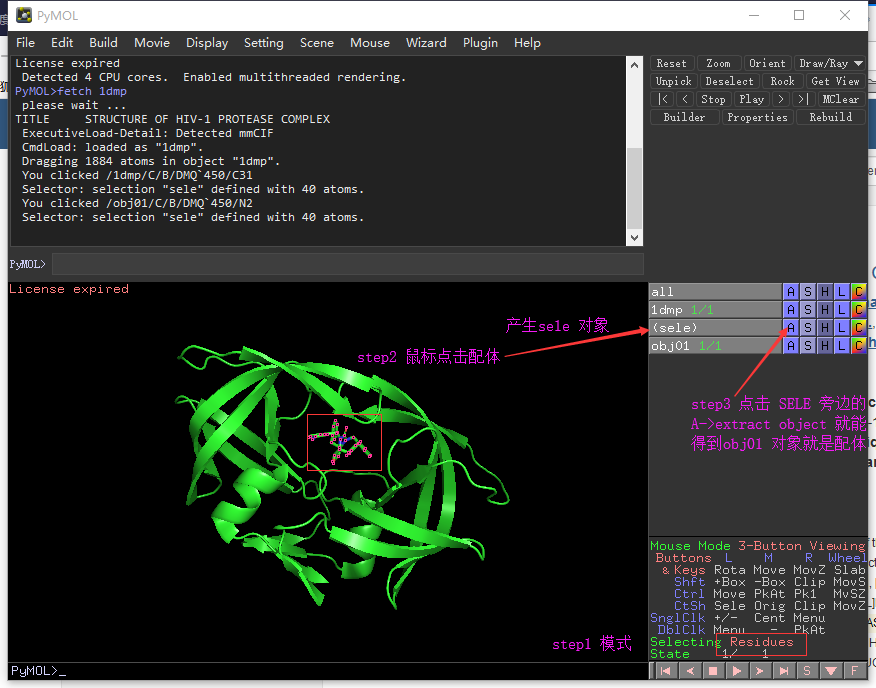
2.2.1 附:用 PyMol 导出 pdb 文件中的配体的方法
3 对接
视频中的步骤:
1、导入protein蛋白质
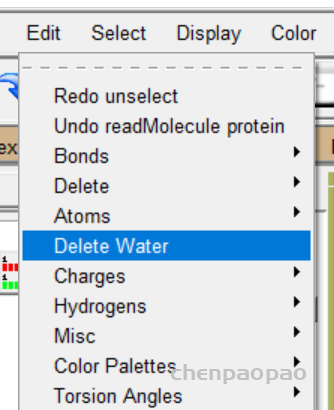
2、删除水分子 如果你的对接区域有水分子,会影响对接结果
pdq该格式省略了一切氢原子,包括游离的水分子都只保留了一个 O,所以后续需要加氢操作。
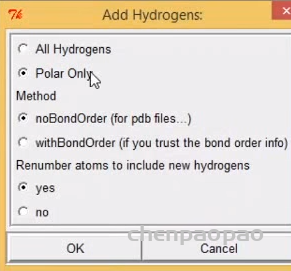
3、edit ->hydrogens->add->polar only 此时结构中发亮的就是氢键(加氢)
4、加电荷 edit -> charges->add kollman charges
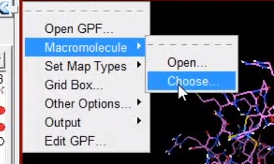
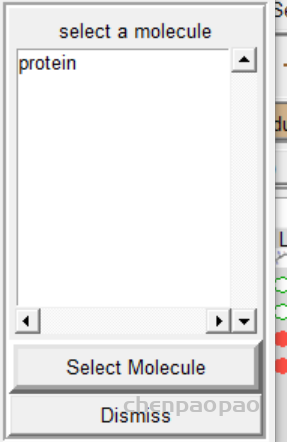
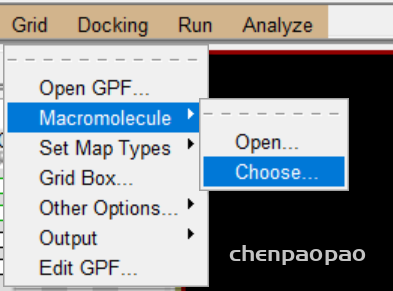
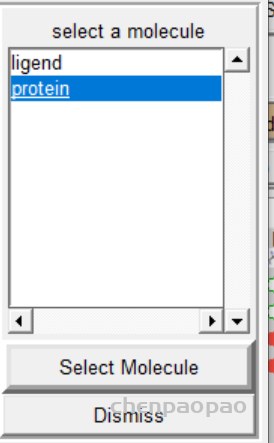
5 报存 grid -> macromolecule ->choose->select
准备配体文件:
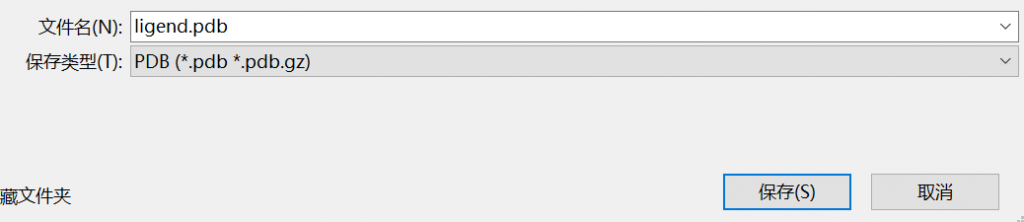
以sdf结尾的文件直接拖进 AUTO软件中会报错,需要转换成pdb文件
可以使用pymol可视化工具转换(注意:配体 英文 ligend)
1、将该文件拖动到pymol中打开,file ->molecule
配体文件:
1、打开auto dock,将配体文件导入:
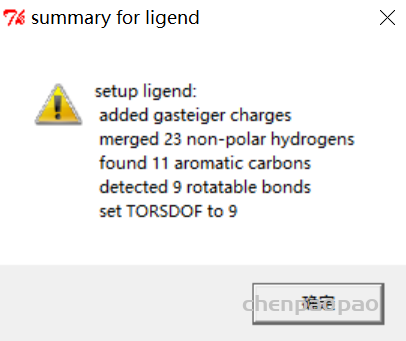
2、 点击 Ligand(配体)->input->choose (这一步就是生成了配体文件)
3、点击 Ligand(配体)->output->save as pdbqt
接下来就可以进行分子对接:
1、将两个(受体和配体)pdpqt导入
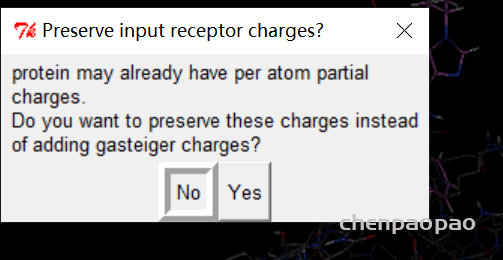
2、重新选择蛋白质分子作为受体
点击NO
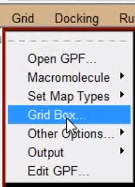
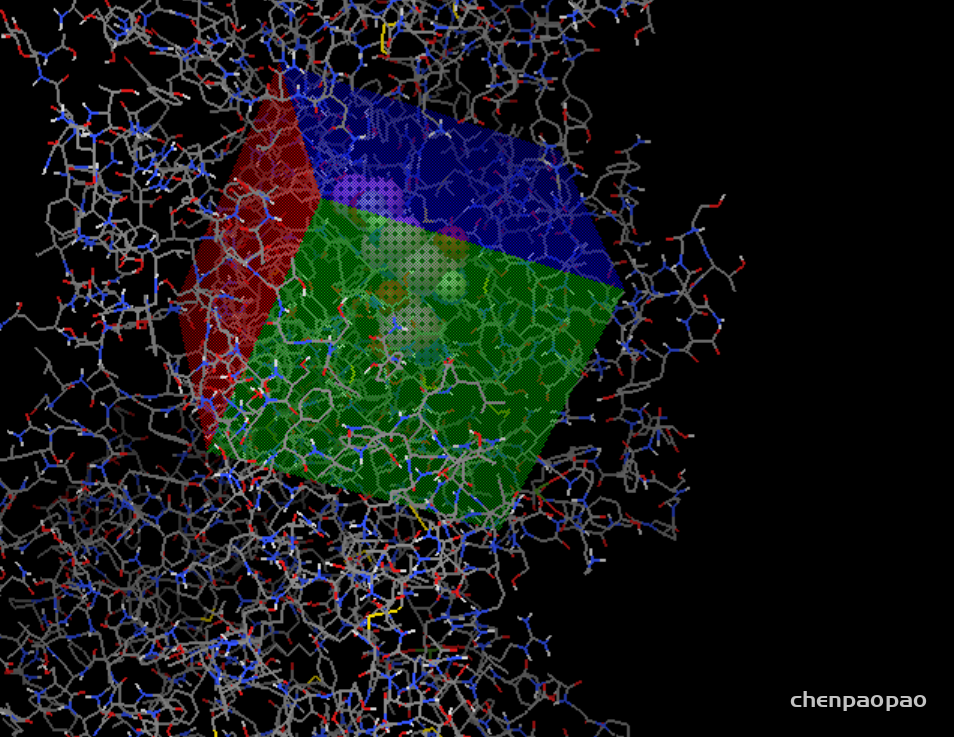
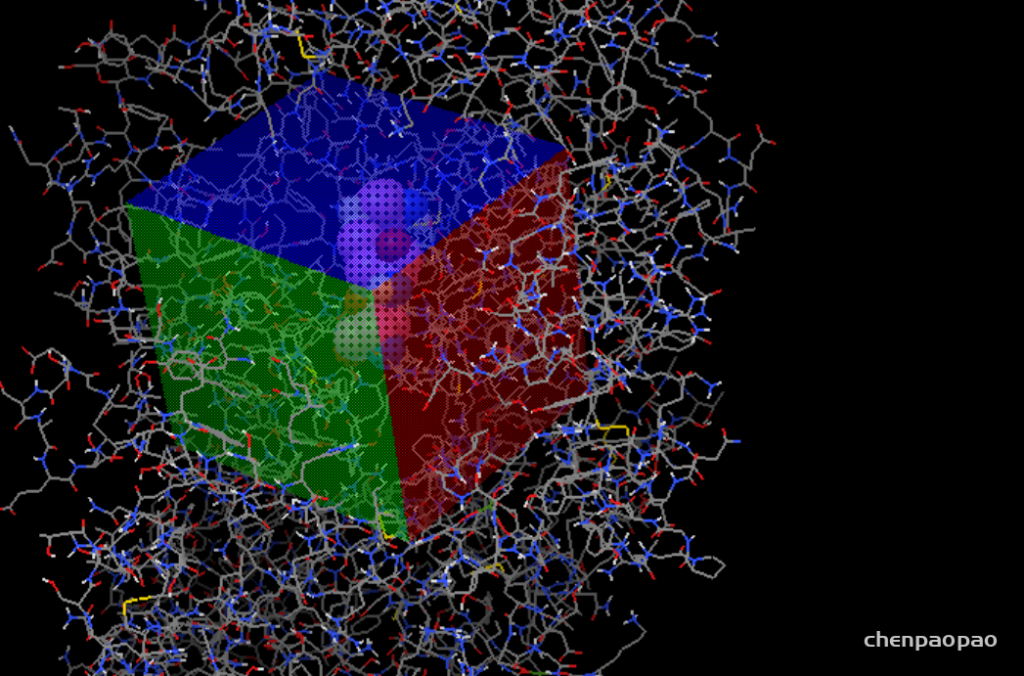
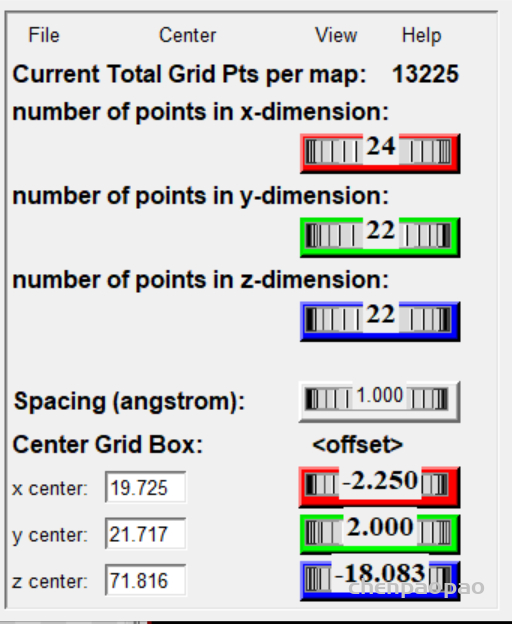
接下来设置对接盒子:
grid -> grid box
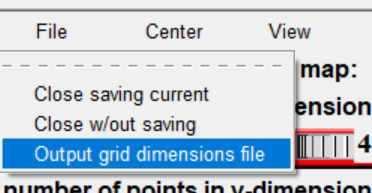
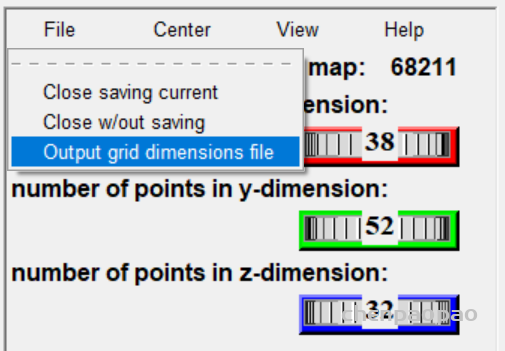
设置盒子位置(spacing设置为1)
然后 grid box 弹出设置中选择 file->output grid dimensions file(保存盒子设置)
保存:
新建config.txt:用于启动vina
receptor 蛋白质名(生成的蛋白质文件名)
ligand 配体名
center 和size在上一步的grid中有
receptor:指定受体分子的路径
ligand:配体分子的路径
center_x,center_y,center_z:搜索空间中心的坐标
size_x,size_y,size_z:指定搜索空间的大小。这里设置的大小基本就是把整个受体分子都包含了,属于blind docking。如何更准确确定结合口袋的位置,我们稍后再说。
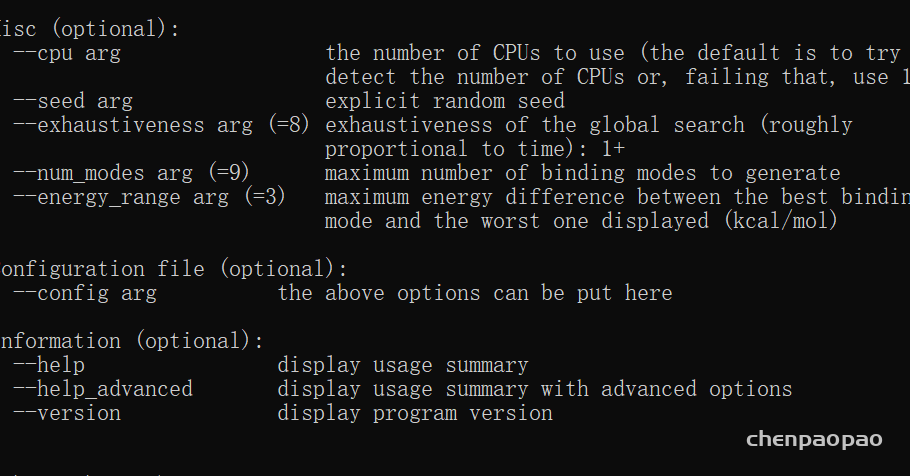
energy_range:默认4,与最优结合模型相差的最大能量值,单位是kcal/mol。比方说,最优模型的能量计算出来是-8.5kcal/mol,那么vina也就最多计算到-4.5kcal/mol的模型就终止了,也就意味着这个值决定了生成模型的最大个数。
exhaustiveness:用来控制对接的细致程度,默认值是8. 大致与时间成正比。
num_modes:最多生成多少个模型。实际生成的模型数由num_modes和energy_range共同决定。
energy_range
maximum energy difference between the best binding
mode and the worst one displayed (kcal/mol)
最后启动vina 进行分子对接:
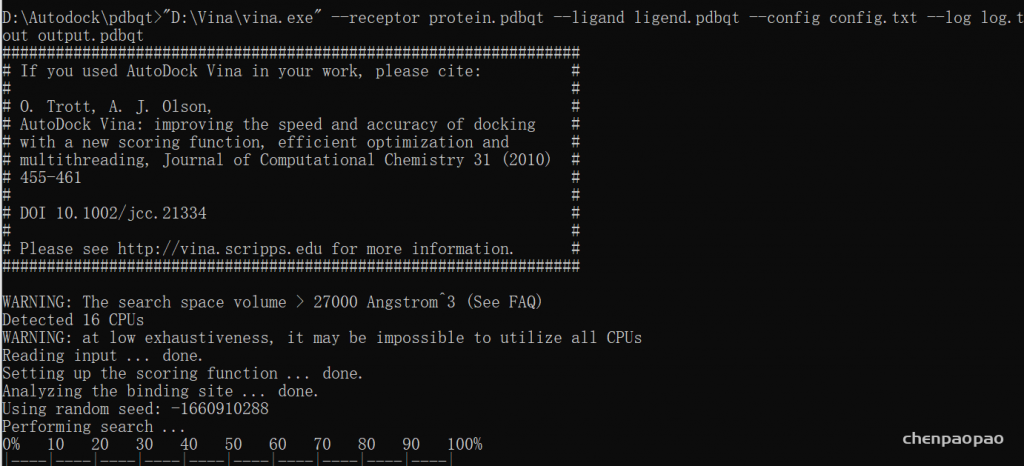
在config目录中打开cmd->输入vina
其中:这条命令就是利用config.txt文件进行分子对接(cmd 必须在config文件目录下打开)
执行 命令:
D:\Autodock\pdbqt>”D:\Vina\vina.exe” –receptor protein.pdbqt –ligand ligend.pdbqt –config config.txt –log log.txt –out output.pdbqt
“D:\Vina\vina.exe” –receptor selected_prediction_ready.pdbqt –ligand Conformer3D_CID_65536.pdbqt –config config.txt –log log.txt –out output.pdbqt
如果上述命令报错,一般是生成的config文件有问题:
正确的config内容如下:
执行完毕可以看到生成一个log文件
vina 中的affinity是亲合力结果排名
最后:
查看生成的模型:
将protein和output输出导入pymol
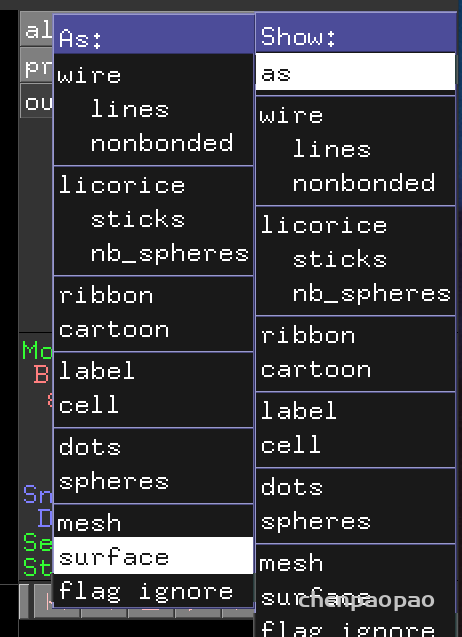
点击 all -> s查看表面 结构
点击左右箭头查看不同的结构:
5.我们挑选第一个模型,看看结构方式是怎样的,见下图。很显然与真实的结合方式相差甚远,可以说是完全错误。
为什么会出现这种情况,很大程度上是因为search space太大,可能需要设置更大的exhaustiveness。
如果我们大致知道binding pocket在什么位置,那准确性应该会高不少,如何大致确定binding pocket的位置呢?我们接着试验。
可以通过实验的方式,比如某个点突变对结合或者活性影响非常大,那么大概率这个残基是结合口袋的一部分。
可以通过软件预测,比如蛋白与配体(底物)结合位点预测:https://zhanglab.ccmb.med.umich.edu/COACH/
再比如Discovery studio软件(专业版的),可以很方便的根据受体分子的表面形状来预测结合口袋位置。
口袋的坐标为:
34.3356,14.9412,26.9615
我们修改下对接参数,新的参数如下:
receptor = r.pdbqt
ligand = nap.pdbqt
center_x = 34.3356
center_y = 14.9412
center_z = 26.9615
size_x = 30.0
size_y = 30.0
size_z = 30.0
energy_range = 4
exhaustiveness = 10
num_modes = 10
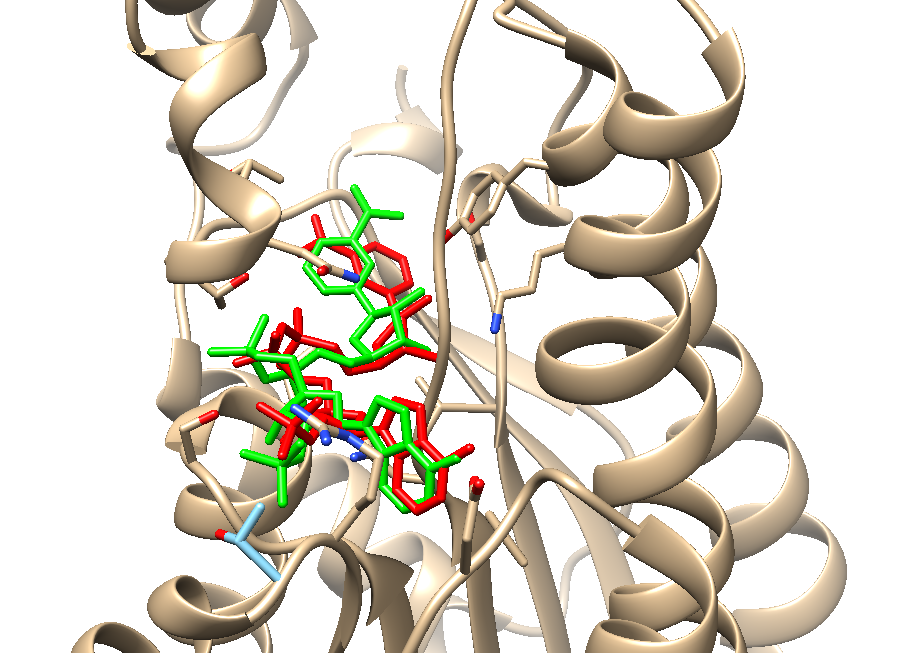
最优结果与真实模型的RMSD为1.795埃,可以说非常精准了
比对结果如下:
Souce: 纽普生物 2019-05-07
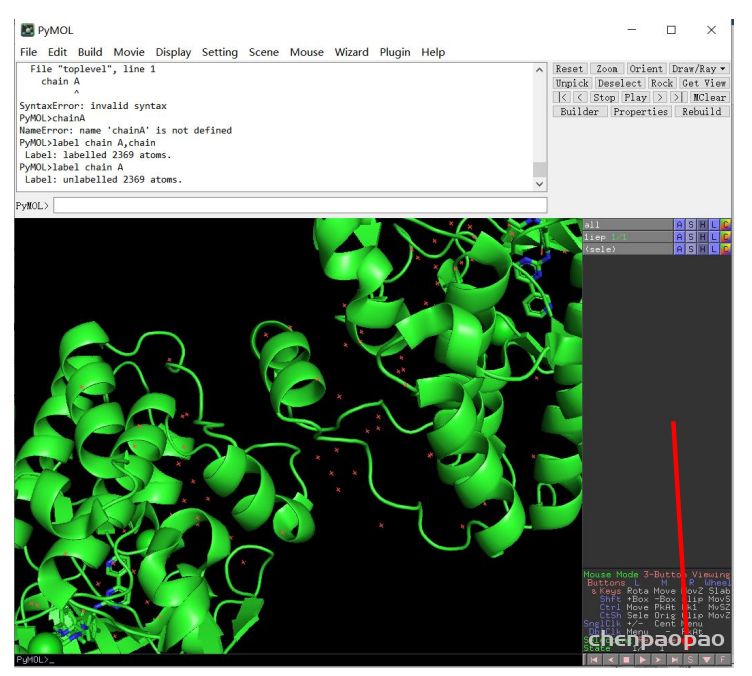
PyMOL 相关操作:
1、导入蛋白质
先在NCBI子数据库structure检索所需要的蛋白结构,https://www.ncbi.nlm.nih.gov/structure/?term=
fetch 5ocn#foxn1
fetch 3uf0#
fetch 1si4#血红蛋白去除水分子
remove solvent
分离得到蛋白
remove organic分离配体:
pymol教程:
http://pymol.chenzhaoqiang.com/intro/startManual.html
分子对接进阶教程:选定对接位点区域:
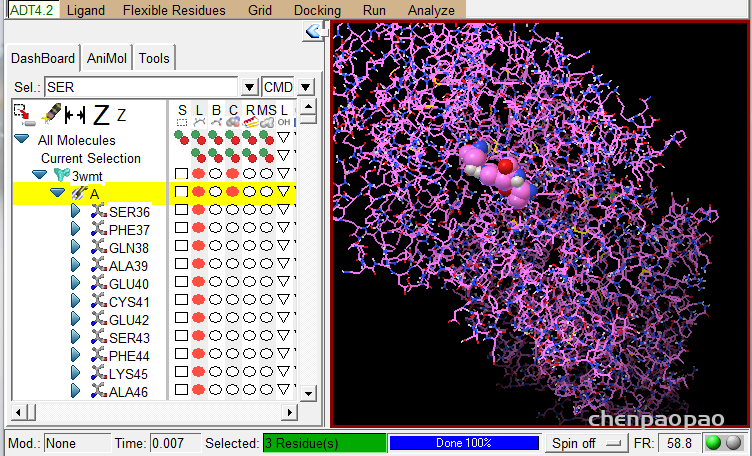
如果我们想要将配体和蛋白质受体的某几个位点部位对接:
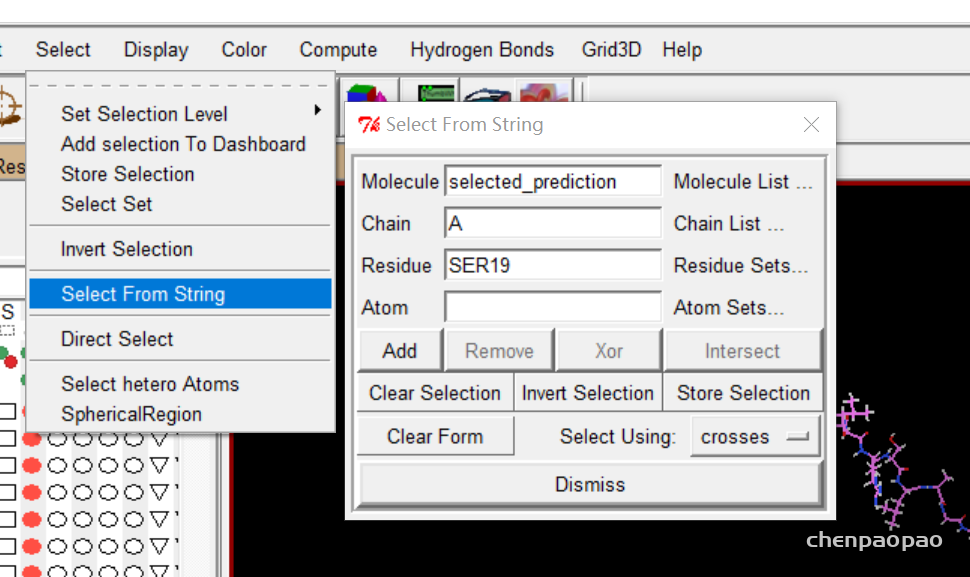
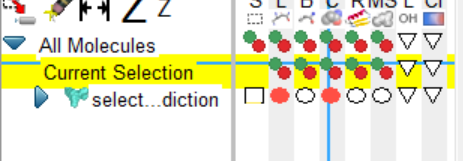
1、标记这些点 select->select from string ,选择对应的molecule和链,以及residue(寻找的位点),点击add
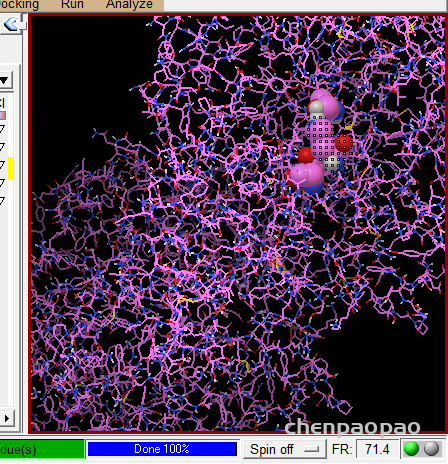
2、此时,会出现currenr selection ,选择该列中 c,点击绿点,会将对应的分子三D化。
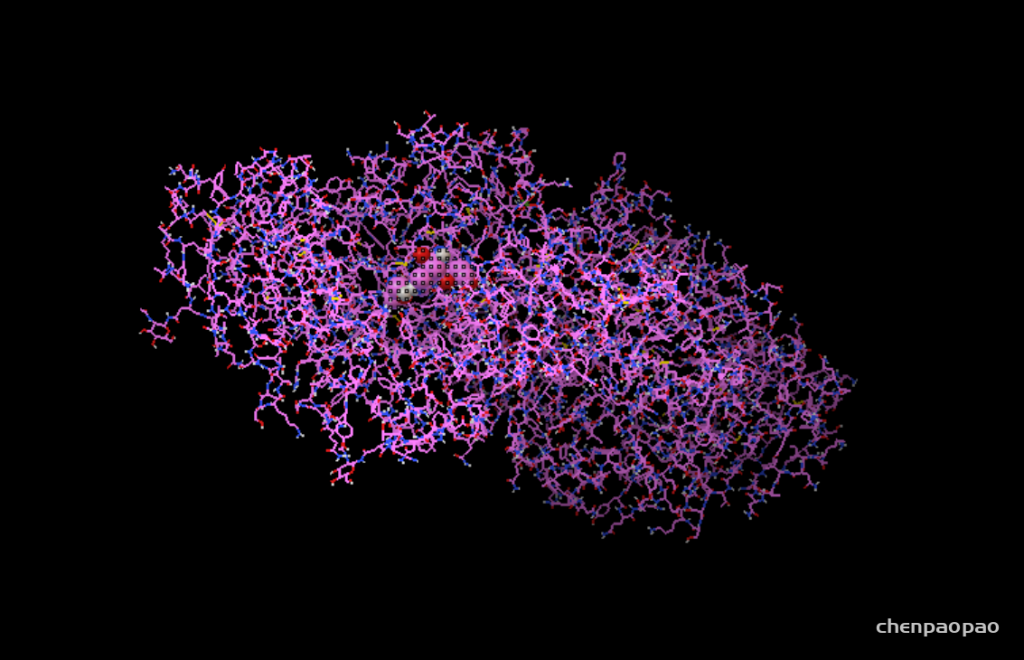
3、接下来设置盒子,spacing 设置为1(相当于比例尺),xyz一般设置为20-22
这样就可以了。